Introduction
Les effets des substituants constituent un concept clé pour la compréhension de la réactivité et du comportement spectroscopique des composés organiques (Krygowski & St??pień, 2005). Dans une approche simple, les effets de substituants peuvent être classés selon le mécanisme d’interaction avec le centre réactif comme des effets inductifs (via des liaisons σ) ou de résonance (via des liaisons π)., Néanmoins, d’autres termes (tels que les effets stériques, sur le terrain ou au solvant) seraient nécessaires pour une description approfondie des effets des substituants.
Depuis la classification D’Ingold des effets des substituants électroniques (Ingold, 1953), le groupe alkyle a été considéré comme un substituant σ-donneur (+I, dans la nomenclature D’Ingold) dans la plupart des manuels de chimie organique (Burrows, Holman, Parsons, Pilling, & Price, 2013; Hornback, 2006; Roos & Roos, 2014; Smith, 2013; vollhardt & Schore, 2014)., Néanmoins, les critiques de L’Eğe à un point de vue aussi simpliste doivent être remarquées:
« dans l’eau, l’acide propanoïque est légèrement plus faible que l’acide acétique. La nature de l’effet inductif d’un groupe alkyle est débattue par les chimistes. Les groupes alkyles stabilisent les carbocations et, dans ce rôle, semblent libérer des électrons. Ils augmentent également la basicité des amines, suggérant à nouveau qu’elles libèrent des électrons. D’autre part, bien que l’alcool tert-butylique (pKa 19) soit un acide plus faible que l’éthanol (PKA 17) dans l’eau, il est un acide plus fort en phase gazeuse., Cette observation expérimentale suggère que les groupes alkyles peuvent stabiliser les anions ainsi que les cations et que la solvatation joue un rôle important dans la détermination des acidités relatives. Donc un mot de prudence est nécessaire. Les acidités relatives sur lesquelles reposent les généralisations présentées dans ce chapitre ont été déterminées dans l’eau. En phase gazeuse, des inversions de l’ordre des composés apparentés sont souvent observées. »(Eğe, 1999, p. 107)
certains effets de substitution alkyle ont souvent été expliqués dans les manuels de manière contradictoire ou énigmatique. , Ainsi, les différences de déplacement chimique entre les groupes CH3 et CH2 sont attribuées dans le Livre de Hornback au fait que « le carbone est légèrement plus électronégatif que l’hydrogène” (Hornback, 2006, p. 549) bien que le groupe alkyle ait été précédemment classé comme un substituant inductif faible donnant des électrons (Hornback, 2006, p. 117). Dans le manuel de Vollhardt, la relation entre les déplacements chimiques du groupe méthyle pour un certain nombre de composés CH3X et L’électronégativité X est illustrée par un tableau dépourvu d’entrée pour X = méthyle (Vollhardt & Schore, 2014, p., 389), évitant ainsi le problème gênant du carbone.
je montre ici que le groupe alkyle se comporte comme un substituant –I+R. Bien que certains facteurs (tels que les effets de champ, stériques ou de solvant) soient implicitement ignorés dans cette approche, de nombreuses preuves théoriques et expérimentales actuellement disponibles peuvent ainsi être décrites de manière simple.
une polarisation de liaison Cδ–Hδ+ a été expérimentalement observée pour le méthane (Lazzeretti, Zanasi,& Raynes, 1987), en cohérence avec l’électronégativité plus grande du carbone par rapport à l’hydrogène, 2,55 contre 2.,20 dans L’échelle de Pauling (Allred, 1961). Un tel schéma de polarisation permet de prédire la direction du moment dipolaire des hydrocarbures simples au moyen de modèles additifs, bien que l’accord quantitatif soit généralement modeste (2-méthylpropane: 0,3 D estimé contre 0,132 D expérimental) (Dean, 1999).
étant donné que l’hydrogène est utilisé comme norme dans la classification des substituants D’Ingold (Krygowski& St??pień, 2005), le groupe alkyle doit être classé comme substituant a –I (d’où un groupe de retrait d’électrons σ). Un tel rôle est illustré à la Fig., 1 pour la polarisation de la liaison σ De n’importe quel atome Y à un groupe méthyle, bien que la polarisation de la liaison inverse soit attendue lorsque Y est plus électronégatif que le carbone (par exemple, le chlore).
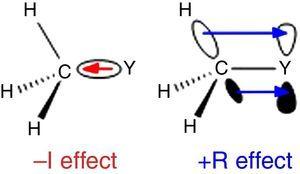
–je (à gauche) et R (droite) les effets d’un groupe méthyle lié à un atome Y.
un comportement différent est trouvé pour les groupes alkyles lorsqu’ils sont attachés à des atomes SP2 ou SP-hybridés en raison du don de densité électronique des liaisons σ alkyles C–H ou C–C à l’orbitale p vide de l’atome contigu (le 1. Ainsi, la diminution de l’acidité en phase gazeuse du phénol et de l’acide benzoïque par substitution p-méthyle (McMahon & Kebarle, 1977) ne peut être attribuée qu’à un effet π-donneur significatif pour le substituant méthyle (en effet, plus important que celui du groupe méthoxy)., Cependant, le groupe alkyle doit être considéré comme un substituant π-donneur atypique en raison de l’absence de paires d’électrons solitaires. Une telle interaction σ-liaison/π-système, appelée hyperconjugation (Mullins, 2012) peut facilement être expliquée par analogie avec le comportement π-donneur d’un atome portant une paire solitaire (par exemple, le chlore) à une orbitale p vide, bien que les liaisons C-C ou C–H (plutôt que les paires solitaires d’électrons) du groupe alkyle soient impliquées en tant, Fait intéressant, les interactions π→σ* (hyperconjugation négative) sont généralement négligeables pour les groupes alkyles dépourvus d’atomes électronégatifs (Bocca, Pontes, & Basso, 2004).
certaines caractéristiques structurales moléculaires peuvent être rationalisées sur la base des propriétés du groupe alkyle. Par exemple, les longueurs de liaison CO plus grandes trouvées dans les méthylcétones (acétone: exp. 1.210 Å, calc. 1.193 Å) en comparaison avec les aldéhydes apparentés (acétaldéhyde: exp. 1.209 Å, calc. 1.,188å) (Berry, Waltman, Pacansky, & Hagler, 1995) peut être attribuée à la stabilisation de la forme de résonance zwitterionique (voir Fig. 2) par le groupe alkyle π-don à l’atome de carbone carbonylique, affaiblissant ainsi la caractéristique de double liaison du groupe carbonyle.
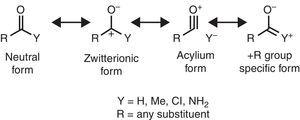
formes de résonance neutre (à gauche) et zwitterionique (à droite) d’un composé carbonyle.
les interactions Hyperconjugatives dépendent de l’arrangement des liaisons C–H (ou C–C) par rapport à l’orbitale p de L’atome y contigu, l’interaction la plus efficace correspondant à un arrangement presque parallèle. Par exemple, la liaison toluène csp3–H presque perpendiculaire au plan du cadre est légèrement plus longue que les autres liaisons csp3–H (de 0,002 Å, Hameka & Jensen, 1996)., La dépendance géométrique de l’hyperconjugation permet d’expliquer l’analyse conformationnelle de composés insaturés méthyl-substitués, tels que le propène (Liberles, O’Leary, Eilers, & Whitman, 1972) ou l’acétaldéhyde (Muñoz-Caro, Niño, & Moule, 1994).
conséquence bien connue du comportement des donneurs π du groupe alkyle, la substitution alkyle donne plus d’alcènes et d’arènes riches en électrons (Libit& Hoffmann, 1974)., La réactivité élevée d’un arène substitué par un alkyle dans une réaction de SEAr peut donc être attribuée à la stabilisation de L’intermédiaire de Wheland correspondant par Don d’électrons π.
le comportement –I+R du groupe alkyle permet d’expliquer un certain nombre de caractéristiques des composés substitués par des alkyles, telles que les moments dipolaires, les propriétés spectroscopiques et la réactivité (en phase gazeuse et en milieu solution), comme indiqué ci-dessous.
moments dipolaires
le comportement de retrait des électrons du groupe alkyle dans les composés aliphatiques se reflète également dans les moments dipolaires., Ainsi, les vecteurs de moment dipolaire pour le propane et le 2-méthylpropane (Tasi et al., 1997), ainsi que certains bicyclooctanes substitués (Böhm & Exner, 2004) peuvent être attribués à l’effet de retrait (- I) du groupe méthyle par rapport à l’hydrogène (voir Fig. 3).
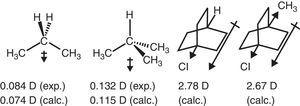
moments dipolaires du propane et des bicyclooctanes substitués.
En revanche, le π-donneur de méthyle du groupe (+R) est nécessaire pour expliquer la hausse des moments dipolaires de nitrobenzène et le benzonitrile par p-méthyl substitution (Brown, 1959) (voir Fig. 4).
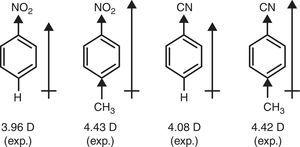
moments dipolaires des dérivés du benzène.
Moléculaire moments dipolaires peut être calculée de manière fiable par le courant des méthodes de calcul., Fait intéressant, les vecteurs de moment dipolaire calculés pour un ensemble d’hydrocarbures simples (Tasi et al., 1997) ont permis de déduire un double rôle pour le groupe méthyle: retrait d’électrons lorsqu’il est attaché à des atomes de carbone sp3, mais don d’électrons lorsqu’il est lié à des carbones sp2 ou sp3.
un tel comportement dual du substituant alkyle est également observé pour les composés portant des hétéroatomes. Ainsi, une diminution progressive du moment dipolaire est observée pour la substitution méthylique successive sur l’ammoniac (NH3, 1,47 D; MeNH2, 1,31 d; Me2NH, 1,01 d; Me3N, 0.,61 D) (Le Fèvre & Russell, 1947), en accord avec la diminution progressive de la densité électronique de l’azote (Hehre & Pople, 1970). En revanche, une amélioration du moment dipolaire (de 1,53 D à 1,68 D) (Nelson, Lide, & Maryott, 1967) est trouvée pour la substitution n,N-diméthyle sur l’aniline (Targema, Obi-Egbedi, & Adeoye, 2013), en cohérence avec l’augmentation de la caractère donneur pour le groupe amino (Hinchliffe & Kidd, 1980) en raison des contributions +r des substituants méthyles.,
propriétés spectroscopiques
Les propriétés spectroscopiques de nombreux composés organiques peuvent être facilement rationalisées en supposant un comportement a –I+R pour le groupe alkyle comme caractéristique générale. Ainsi, le déplacement chimique RMN d’un atome peut être considéré comme une mesure expérimentale de la densité électronique à la position correspondante du noyau bien que d’autres effets – tels que les champs magnétiques anisotropes – puissent également être impliqués. Champ faible déplace induite par un substituant méthyle sur le service pack 3 atomes de carbone (de+9,6 ppm en RMN 13C) ou des correspondants liés aux atomes d’hydrogène (+0.,63ppm dans la RMN du 1H) (Pretsch, Bühlmann, & Badertscher, 2009) sont compatibles avec le comportement des groupes –I typiques (tels que les atomes d’halogène).
Les effets de substitution des alkyles sur les déplacements chimiques des alcènes en RMN montrent une diminution de la densité électronique en position α (+12,9 ppm pour la RMN du 13C; +0,45 ppm pour la RMN du 1h), ainsi qu’une augmentation de la densité en position β (-7,4 ppm pour le 13c; -0,31/-0,40 ppm pour le 1h), en cohérence avec l’effet a –I+R, Bien que des effets anisotropes (tels que des courants cycliques) puissent également jouer un rôle. Un tel comportement-I + R est également trouvé pour les alcynes, selon la spectroscopie RMN 13C (+8.,5 ppm pour la position α, -3,6 ppm pour la position β).
le comportement dichotomique des substituants alkyles sur les systèmes π (augmentation de la densité électronique pour l’atome α, diminution de la densité électronique pour l’atome β) ne peut pas être expliqué sur la base d’un comportement simple (tel que l’effet a +I).
le comportement A –I+R (Meier, 2007) est observé par spectroscopie RMN du 15N pour la substitution alkyle sur les amines et les amides en fonction de l’hybridation de l’azote (décalages vers le bas pour les amines aliphatiques, décalages vers le haut pour les composés nsp2-tels que les anilines et les amides).,
Les constantes de couplage RMN dépendent également des propriétés électroniques des substituants (ainsi que de certaines caractéristiques géométriques). Ainsi, une diminution significative est trouvée pour les constantes de couplage 1H–1H par substitution méthylique (trans, -2,3 Hz; cis, -1,6 Hz; gem, -0,4 Hz), en accord qualitatif avec les données des groupes électron-retirants typiques, tels que l’atome de fluor (trans, -6,3 Hz; cis, -6,9 Hz; gem, -5,7 Hz). La contribution positive pour le méthyl-substitution sur 13C–1H les constantes de couplage des composés aliphatiques (+1.,0Hz), est également qualitativement compatible avec ceux des autres groupes –I (fluor, +24Hz).
la spectroscopie infrarouge est également sensible aux propriétés des substituants, comme l’illustre la fréquence D’étirement CO des composés carbonyles en fonction du substituant y correspondant, qui peut être rationalisée en termes de formes de résonance (Fig. 2). En prenant un aldéhyde aliphatique (env., 1725cm-1) à titre de référence, le décalage vers le rouge (diminution du nombre d’ondes) induit par un substituant a +I (acétyltriméthylsilane, 1645cm–1: Soderquist & Hsu, 1982) peut être attribué à la stabilisation de la forme zwitterionique. Au lieu de cela, le blueshift provoqué par un substituant a-I (chlorures d’acyle, > 1800cm–1: Pretsch et al., 2009) peuvent s’expliquer au moyen de deux mécanismes alternatifs ou simultanés (déstabilisation de la forme zwitterionique et/ou contribution d’une forme porteuse d’ions acylium). Enfin, les redshifts provoqués par les substituants + R (amides, ca., 1680cm–1: Pretsch et coll., 2009) peut être attribuée à la contribution d’une forme de résonance spécifique. Le léger décalage vers le rouge induit par le groupe alkyle (méthylcétones, ca. 1715cm-1) montre un effet net de Don d’électrons (d’où une prédominance de l’effet +R sur les propriétés-I). L’effet donneur net du groupe alkyle lié au carbonyle est compatible avec le plus grand moment dipolaire de l’acétone (2,88 D) par rapport au formaldéhyde (2,33 d) (Nelson et al., 1967).
l’influence du groupe alkyle sur les spectres UV–Vis de nombreux composés peut également être expliquée en termes d’effets électroniques., Ainsi, les déplacements bathochromiques induits par les groupes alkyles sur les bandes D’absorption UV des composés α, β-insaturés (+10 nm en position α, +12 nm en position β), Des polyènes conjugués (+5 nm) ou des dérivés benzéniques (+3,0 nm) sont qualitativement compatibles avec les effets des groupes π-donneurs typiques (par exemple, le chlore).
réactivité acide–base en phase gazeuse
Les basicités relatives des amines aliphatiques en solution aqueuse ont été attribuées à l’effet supposé +I du groupe alkyle (Sorrell, 2006)., Interestingly, the irregular basicity order of amines in water (Me2NH>MeNH2>Me3N>NH3, as shown by the pKa values for the corresponding conjugated acids: 10.77>10.62>9.80>9.,246) (Dean, 1999) est contaminée par les effets des solvants comme l’illustre l’ordre de basicité systématique des amines en phase gazeuse (Me3N>Me2NH>MeNH2>NH3) (Brauman, Riveros, & Blair, 1971)., Bien que l’ordre de basicité en phase gazeuse puisse être attribué à l’effet alkyle +I habituellement supposé (Carter, 2007), une diminution de la densité électronique de l’azote par substitution méthylique a en effet été observée au moyen de calculs de potentiel électrostatique moléculaire (Baeten, de Proft, & Geerlings, 1995), indiquant ainsi un comportement a –I pour le groupe méthyle., En fait, l’ordre de basicité en phase gazeuse des amines aliphatiques doit être attribué à la stabilisation croissante des ions ammonium substitués due à la polarisabilité du groupe alkyle (Aue, Webb, & Bowers, 1976).
les acidités relatives des alcools en solution aqueuse (H2O>MeOH>EtOH>iPrOH>tBuOH) ont également été attribuées manuels sur l’effet supposé alkyle +I (Johnson, 1999; Solomons, fryhle, & Snyder, 2016)., Puisque l’ordre d’acidité inverse se trouve en phase gazeuse, les acidités relatives des alcools dans l’eau doivent être attribuées aux magnitudes plus faibles des enthalpies de solvatation pour les anions alkoxydes plus gros (Brauman & Blair, 1969).
la discussion sur les propriétés électroniques des groupes alkyles peut également être appliquée aux carbanions. Ainsi, l’ordre de stabilité « manuel » des carbanions simples (méthyl >éthyle>isopropyle> tert-butyle) a été attribué à l’effet inductif supposé +I des groupes alkyles (Burrows et al.,, 2013; Chaloner, 2015; Roos & Roos, 2014; Smith, 2013). Cependant, un ordre irrégulier est trouvé pour les stabilités des carbanions en phase gazeuse (tBu>Me>iPr> Et), en accord avec la concomitance de deux effets alkyles opposés (DePuy et al., 1989): un mécanisme stabilisant par polarisabilité alkyle (c’est-à-dire, N→σ* hyperconjugation) et une tendance déstabilisante (toujours avec un rôle a +R, en supposant un comportement de type p pour la paire solitaire de carbone).,
la stabilité d’autres intermédiaires réactionnels peut également être évaluée sur la base des effets du groupe alkyle. Ainsi, l’ordre de stabilité bien connu des carbocations (tertiaire>secondaire>primaire>méthyle) a parfois été attribué à un effet inductif positif (Chaloner, 2015; Roos & Roos, 2014)., Fait intéressant, l’hyperconjugation est présentée dans de nombreux manuels comme une explication alternative de l’ordre de stabilité des carbocations (Brown ,verson, Anslyn, & Foote, 2013; Burrows et al., 2013) bien que l’écriture ambiguë habituelle empêche de déterminer si les deux explications correspondent à deux descriptions différentes du même phénomène ou à deux mécanismes simultanés jouant dans la même direction., Quoi qu’il en soit, l’ordre de stabilité des carbocations doit être attribué à l’hyperconjugation (d’où le comportement a+R sur une orbitale P vacante, le système π le plus simple), bien que d’autres interactions (telles que la polarisabilité des alkyles) soient également impliquées (Aue, 2011).
les radicaux libres présentent le même ordre de stabilité que les carbocations, indiquant ainsi une stabilisation par substitution alkyle. Bien qu’un tel ordre de stabilité puisse être justifié sur la base d’un comportement supposé +I, l’effet +R peut être considéré alternativement, de manière analogue à la stabilisation des radicaux libres par des atomes porteurs de paires solitaires (Zipse, 2006).,
réactivité en solution
les acidités relatives des acides carboxyliques simples en solution aqueuse (acide acétique>acide propionique>acide butyrique) ont été utilisées dans certains manuels pour illustrer l’effet supposé +I du groupe alkyle (Sorrell, 2006). Fait intéressant, l’ordre inverse est trouvé lorsque les enthalpies sont plutôt considérées (Christensen, Izatt, & Hansen, 1967), indiquant ainsi que l’ordre de l’acidité en solution aqueuse doit être attribué aux entropies d’hydratation., Ainsi, l’ordre de réseau significatif de l’eau liquide (entropie de vaporisation égale à 118,89 Jmol–1K-1, contrairement aux valeurs typiques de ca. 88jmol–1K-1 pour la plupart des liquides, Dean, 1999) peut introduire des changements importants sur l’énergie de réaction. En particulier, l’hydratation des molécules apolaires (ou des fractions) conduit à un autre ordre de réseau de solvants (Blokzijl & Engberts, 1993). En conséquence, les effets inductifs du groupe alkyle provenant de données expérimentales en solution aqueuse sont souvent masqués par des entropies d’hydratation (Calder & Barton, 1971)., Acidités relatives des acides carboxyliques simples en phase gazeuse (Yamdagni & Kebarle, 1973) et de l’acétonitrile (Eckert et al., 2009) sont compatibles avec le rôle majeur joué par les entropies d’hydratation.
la plus faible acidité de l’acide pivalique par rapport à l’acide acétique, généralement attribuée à l’effet supposé +I du groupe alkyle (Smith, 2008), est inversée lorsque l’on considère les enthalpies de réaction (Eckert et al., 2009).,
l’effet supposé du groupe alkyle +I sur l’acidité des acides carboxyliques simples en solution aqueuse peut donc être attribué à un artefact dérivé des effets du solvant. Alors qu’une augmentation du volume des solutés neutres conduit à une augmentation de l’entropie d’hydratation, la relation inverse est trouvée pour les espèces ioniques (Graziano, 2009). En conséquence, la substitution alkyle (par une augmentation du volume moléculaire) conduit à la stabilisation (en termes D’énergie libre de Gibbs) de l’acide non ionisé dans l’eau ainsi qu’à la déstabilisation de l’anion carboxylate correspondant, entraînant ainsi une diminution de l’acidité.,
La plus grande acidité de l’acide formique en comparaison avec de l’acide acétique en solution aqueuse (valeurs pKa: 3.751 et 4.756, respectivement, Dean, 1999) a également été discutée dans de nombreux manuels scolaires comme un exemple de l’application des effets inductifs (Hart, Hadad, Craine, & Hart, 2012; Hornback, 2006; Okuyama & Maskill, 2014; Roos & Roos, 2014). Étant donné que des enthalpies de réaction très similaires sont impliquées dans les réactions de dissociation des acides formique et acétique (Christensen et al.,, 1967), la plus grande acidité de l’acide formique doit en effet être attribuée aux différences d’entropie d’hydratation.
Conclusions
une compréhension claire des effets inductifs et de résonance est une clé majeure pour un apprentissage sain de la chimie organique (Mullins, 2008). Étonnamment, le groupe alkyle presque omniprésent a été incorrectement présenté dans de nombreux manuels comme un groupe σ-donneur (+I). Cependant, un comportement dual est montré par les substituants alkyles en fonction de l’hybridation de l’atome voisin., Ainsi, les groupes alkyles liés aux chaînes aliphatiques se comportent comme des σ-accepteurs (–I, en cohérence avec l’électronégativité plus grande du carbone par rapport à l’hydrogène), tandis que ceux attachés aux systèmes π agissent comme des π-donneurs (+R, en raison d’interactions hyperconjugatives). Un certain nombre de données expérimentales et théoriques (moments dipolaires, RMN, spectres IR et UV, réactivité) concordent avec un tel comportement dual.,
l’ensemble de l’analyse de toutes les données considérées ici permet d’inférer un petit effet –I ainsi qu’un comportement significatif +R pour le groupe alkyle en tant que caractéristique valable dans toutes les discussions sur les propriétés spectroscopiques et de réactivité des composés organiques.
Conflit d’intérêts
L’auteur ne déclare aucun conflit d’intérêt.