Introducción
Los efectos sustituyentes constituyen un concepto clave para la comprensión de la reactividad y el comportamiento espectroscópico de los compuestos orgánicos (Krygowski & St??pień, 2005). En un enfoque simple, los efectos sustituyentes se pueden clasificar según el mecanismo de interacción con el centro reactivo como inductivos (a través de enlaces σ) o efectos de resonancia (a través de enlaces π)., Sin embargo, se requerirían algunos términos adicionales (como efectos estéricos, de campo o solventes) para una descripción exhaustiva de los efectos sustituyentes.
desde la clasificación de Ingold de los efectos sustituyentes electrónicos (Ingold, 1953), el grupo alquilo ha sido considerado como un sustituyente σ-donante (+I, en la nomenclatura de Ingold) en la mayoría de los libros de texto de Química Orgánica (Burrows, Holman, Parsons, Pilling, & Price, 2013; Hornback, 2006; Roos & Roos, 2014; Smith, 2013; vollhardt & Schore, 2014)., Sin embargo, las críticas de la Eğe a un punto de vista tan simplista deben ser remarcadas:
«en el agua, el ácido propanoico es ligeramente más débil que el ácido acético. La naturaleza del efecto inductivo de un grupo alquilo es debatida por los químicos. Los grupos alquilos estabilizan las carbocaciones y en ese papel parecen ser liberadores de electrones. También aumentan la basicidad de las aminas, sugiriendo de nuevo que son liberadoras de electrones. Por otro lado, aunque el alcohol terc-butílico (PKA 19) es un ácido más débil que el etanol (PKA 17) en el agua, es un ácido más fuerte en la fase gaseosa., Esta observación experimental sugiere que los grupos alquilos pueden estabilizar aniones y cationes y que la solvatación juega un papel importante en la determinación de las acididades relativas. Por lo tanto, una palabra de precaución es necesaria. Las acididades relativas en las que se basan las generalizaciones presentadas en este capítulo se determinaron en agua. En la fase gaseosa, a menudo se ven reversiones en el orden de los compuestos relacionados.»(Eğe, 1999, p. 107)
algunos efectos de sustitución de alquilos se han explicado a menudo en los libros de texto de manera contradictoria o enigmática. , Por lo tanto, las diferencias de desplazamiento químico entre los grupos CH3 y CH2 se atribuyen en el libro de Hornback al hecho de que «el carbono es ligeramente más electronegativo que el hidrógeno» (Hornback, 2006, p. 549) a pesar de que el grupo alquilo ha sido clasificado previamente como un sustituyente inductivo débil que dona electrones (Hornback, 2006, p. 117). En el libro de texto de Vollhardt, la relación entre los cambios químicos del grupo metilo para un número de compuestos CH3X y la electronegatividad X se ilustra mediante una tabla que carece de una entrada para X = metilo (Vollhardt & Schore, 2014, p., 389), evitando así la incómoda cuestión del carbono.
Muestro aquí que el grupo alquilo se comporta como sustituyente a-i + R. Aunque algunos factores (como los efectos de campo, estéricos o solventes) se ignoran implícitamente en este enfoque, muchas de las evidencias teóricas y experimentales actualmente disponibles pueden describirse de una manera fácil.
se ha observado experimentalmente una polarización del enlace cδ H Hδ+ para el metano (Lazzeretti, Zanasi, & Raynes, 1987), consistentemente con la electronegatividad más grande del carbono en relación con el hidrógeno, 2.55 vs.2.,20 en la escala de Pauling (Allred, 1961). Tal patrón de polarización permite predecir la dirección del momento dipolar de hidrocarburos simples a través de modelos aditivos, aunque el Acuerdo cuantitativo es generalmente modesto (2-metilpropano: 0.3 D estimado vs.0.132 D experimental) (Dean, 1999).
dado que el hidrógeno se utiliza como el estándar en la clasificación de sustituyentes de Ingold (Krygowski & St??pień, 2005), el grupo alquilo debe clasificarse como un sustituyente a –I (de ahí un grupo σ de retiro de electrones). Tal papel se ilustra en la Fig., 1 para la polarización del enlace σ de cualquier átomo y a un grupo metilo, aunque se espera la polarización inversa del enlace cuando Y es más electronegativa que el carbono (por ejemplo, cloro).
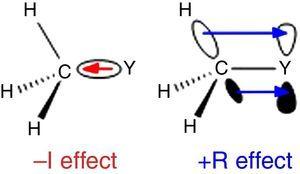
–I (izquierda) y +R (derecha) efectos de un grupo metilo unido a un átomo Y.
se encuentra un comportamiento diferente para los grupos alquilos cuando se unen a átomos SP2 o SP hibridizados debido a la donación de densidad electrónica de los enlaces alquil C–H o c–c σ Al orbital P vacío del átomo contiguo (el sistema π más simple), como se muestra en la Fig. 1. Por lo tanto, la disminución de la acidez en fase gaseosa para el fenol y el ácido benzoico a través de la sustitución de p-metilo (McMahon & Kebarle, 1977) solo puede atribuirse a un efecto significativo del donante π para el sustituyente metilo (de hecho, más grande que el del grupo metoxi)., Sin embargo, el grupo alquilo debe ser considerado como un sustituyente atípico del donante π debido a la falta de pares de electrones solitarios. Tal interacción de enlace σ / sistema π, llamada hiperconjugación (Mullins, 2012) puede explicarse fácilmente por analogía con el comportamiento del donante π de un átomo portador de par solitario (por ejemplo, cloro) a un orbital P vacío, aunque los enlaces C-C O C-H (en lugar de pares solitarios de electrones) del grupo alquilo están involucrados como unidades liberadoras de electrones en interacciones hiperconjugativas., Curiosamente, las interacciones π→σ* (hiperconjugación negativa) son generalmente insignificantes para grupos alquilos que carecen de átomos electronegativos (Bocca, Pontes, & Basso, 2004).
algunas características estructurales moleculares pueden ser racionalizadas sobre la base de las propiedades del grupo alquilo. Por ejemplo, las longitudes de enlace de CO más grandes que se encuentran en las cetonas metílicas(acetona: exp. 1.210 Å, calc. 1.193 Å) en comparación con los aldehídos relacionados (acetaldehído: exp. 1.209 Å, calc. 1.,188Å) (Berry, Waltman, Pacansky, & Hagler, 1995) se puede atribuir a la estabilización de la zwitteriónicos de resonancia (ver Fig. 2) a través del grupo alquilo π-donación al átomo de carbono carbonílico, debilitando así la característica de doble enlace del grupo carbonilo.
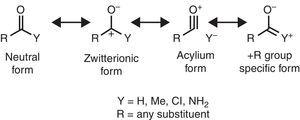
formas de resonancia neutras (izquierda) y zwitterionicas (derecha) de un compuesto carbonilo.
Las interacciones Hiperconjugativas dependen de la disposición de los enlaces C–H (O C–C) En relación con el orbital p del átomo contiguo y, la interacción más efectiva corresponde a una disposición casi paralela. Por ejemplo, el enlace tolueno csp3-H casi perpendicular al plano del marco es ligeramente más largo que los otros enlaces Csp3-H (por 0.002 Å, Hameka & Jensen, 1996)., La dependencia geométrica de la hiperconjugación permite explicar el análisis conformacional de compuestos insaturados sustituidos por metilo, como el propeno (Liberles, O’Leary, Eilers, & Whitman, 1972) o el acetaldehído (Muñoz-Caro, Niño, & Moule, 1994).
como consecuencia bien conocida del comportamiento del donante π del grupo alquilo, la sustitución alquilo produce más alquenos y arenos ricos en electrones (Libit & Hoffmann, 1974)., La alta reactividad de un areno alquilo-sustituido en una reacción de sellado puede atribuirse a la estabilización del intermedio Wheland correspondiente a través de la donación de π-electrones.
el comportamiento-i+R del grupo alquilo permite explicar una serie de características de los compuestos alquilsustituidos, tales como momentos dipolares, propiedades espectroscópicas y reactividad (en fase gaseosa y medios de solución), como se muestra a continuación.
momentos dipolares
el comportamiento de retiro de electrones del grupo alquilo en compuestos alifáticos también se refleja en momentos dipolares., Por lo tanto, los vectores de momento dipolar para propano y 2-metilpropano (Tasi et al., 1997), así como algunos bicyclooctanos sustituidos (Böhm & Exner, 2004) pueden atribuirse al efecto de retirada (–I) del grupo metilo en comparación con el hidrógeno (ver Fig. 3).
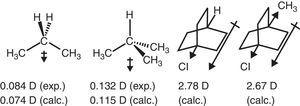
momentos dipolares de propano y bicyclooctanos sustituidos.
en contraste, el carácter π-donante del grupo metilo (+R) se requiere para explicar el aumento de momentos dipolares de nitrobenceno y benzonitrilo a través de la sustitución de P-metilo (Brown, 1959) (ver Fig. 4).
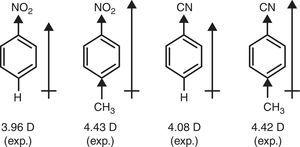
momentos dipolares de derivados del benceno.
Molecular momentos de dipolo puede ser fiable calculado por los actuales métodos de cálculo., Curiosamente, los vectores de momento dipolar calculados para un conjunto de hidrocarburos simples(Tasi et al., 1997) han permitido inferir un doble papel para el grupo metilo: retiro de electrones cuando se une a átomos de carbono sp3, pero donación de electrones cuando se une a átomos de carbono sp2 o sp3.
Este comportamiento dual del sustituyente alquilo también se observa para los compuestos heteroátomos. Por lo tanto, se observa una disminución gradual del momento dipolar para la sustitución sucesiva de metilo en amoníaco (NH3, 1.47 D; MeNH2, 1.31 D; Me2NH, 1.01 D; Me3N, 0.,61 D) (Le Fèvre & Russell, 1947), de acuerdo con la disminución progresiva de la densidad electrónica de nitrógeno (Hehre & Pople, 1970). En contraste, una mejora del momento dipolar (de 1.53 D A 1.68 D) (Nelson, Lide, & Maryott, 1967) se encuentra para la sustitución de N,N-dimetil en anilina (Targema, Obi-Egbedi, & Adeoye, 2013), consistentemente con el aumento del π-carácter donante para el grupo amino (Hinchliffe & Kidd, 1980) debido a contribuciones +R de sustituyentes metilo.,
propiedades espectroscópicas
Las propiedades espectroscópicas de muchos compuestos orgánicos pueden ser fácilmente racionalizadas asumiendo el comportamiento a-i + R para el grupo alquilo como una característica general. Por lo tanto, el desplazamiento químico de RMN de un átomo puede ser considerado como una medida experimental de la densidad electrónica en la posición del núcleo correspondiente, aunque otros efectos, como los campos magnéticos anisotrópicos, también pueden estar involucrados. Desplazamiento descendente inducido por un sustituyente metilo en átomos de carbono sp3 (+9,6 ppm en RMN de 13C) o en los átomos de hidrógeno enlazados correspondientes (+0.,63ppm en 1H NMR) (Pretsch, Bühlmann, & Badertscher, 2009) son consistentes con el comportamiento de los grupos –I típicos (como los átomos de halógeno).
Los efectos de sustitución de alquilo en la RMN los cambios químicos de los alquenos muestran una disminución de la densidad electrónica en la posición α (+12,9 ppm para la RMN de 13C; +0,45 ppm para la RMN de 1H), así como un aumento de la densidad en la posición β (-7,4 ppm para 13C; -0,31/-0,40 ppm para 1H), consistentemente con el efecto a –i+R, aunque los efectos anisotrópicos (como las corrientes de anillo) también pueden desempeñar un papel. Tal comportamiento a-i+R también se encuentra para alquinos, de acuerdo con la espectroscopia de RMN 13C (+8.,5 ppm para la posición α, -3.6 ppm para la posición β).
el comportamiento dicotómico de los sustituyentes alquilos en los sistemas π (aumento de la densidad electrónica para el átomo α, disminución de la densidad electrónica para el átomo β) no puede explicarse sobre la base de un comportamiento simple (como el efecto a +I).
el comportamiento A –i+r (Meier, 2007) se observa a través de la espectroscopia de RMN 15N para la sustitución de alquilo en aminas y amidas dependiendo de la hibridación de nitrógeno (cambios downfield para aminas alifáticas, cambios upfield para compuestos con nsp2-tales como anilinas y amidas).,
Las constantes de acoplamiento de RMN también dependen de las propiedades electrónicas del sustituyente (así como de algunas características geométricas). Por lo tanto, se encuentra una disminución significativa para las constantes de acoplamiento 1H–1h a través de la sustitución de metilo (trans, -2,3 Hz; cis, -1,6 Hz; gem, -0,4 Hz), en concordancia cualitativa con los datos de grupos típicos de retiro de electrones, como el átomo de flúor (trans, -6,3 Hz; cis, -6,9 Hz; gem, -5,7 Hz). The positive contribution for methyl-substitution on 13C-1h coupling constants of alifatic compounds (+1.,0Hz), también es cualitativamente consistente con los de otros grupos –I (flúor, +24Hz).
la espectroscopia infrarroja también es sensible a las propiedades sustituyentes, como lo ilustra la frecuencia de estiramiento de CO de los compuestos carbonílicos en función del sustituyente correspondiente Y, que puede racionalizarse en términos de formas de resonancia (Fig. 2). Tomando un aldehído alifático (ca., 1725cm–1) como referencia, el corrimiento al rojo (disminución del número de onda) inducido por el sustituyente a +I (acetiltrimetilsilano, 1645cm-1: Soderquist & Hsu, 1982) puede atribuirse a la estabilización de la forma zwitterionic. En cambio, el desplazamiento al azul provocado por el sustituyente a –I (cloruros de acil, >1800cm-1: Pretsch et al., 2009) se puede explicar por medio de dos mecanismos alternativos o concurrentes (desestabilización de la forma zwitteriónica y/o contribución de una forma portadora de iones acilio). Finalmente, los corrimientos al rojo provocados por sustituyentes +R(amidas, ca., 1680cm-1: Pretsch et al., 2009) se puede atribuir a la contribución de una forma de resonancia específica. El ligero corrimiento al rojo inducido por el grupo alquilo (cetonas metílicas, ca. 1715cm-1) muestra un efecto neto de donación de electrones (por lo tanto, un predominio de las propiedades del efecto +R sobre –I). El efecto donante neto del grupo alquilo unido al carbonilo es consistente con el momento dipolar más grande de la acetona (2.88 D) en relación con el formaldehído (2.33 D) (Nelson et al., 1967).
la influencia del grupo alquilo en los espectros UV–Vis de muchos compuestos también se puede explicar en términos de efectos electrónicos., Por lo tanto,los cambios batocrómicos inducidos por grupos alquilos en bandas de absorción UV de compuestos α, β-insaturados (+10nm en la posición α, +12nm en la posición β), polienos conjugados (+5nm) o derivados del benceno (+3.0 nm) son cualitativamente consistentes con los efectos de los grupos típicos de donantes π (por ejemplo, cloro).
reactividad ácido–base en fase gaseosa
Las basicidades relativas de las aminas alifáticas en solución acuosa se han atribuido al supuesto efecto +I del grupo alquilo (Sorrell, 2006)., Interestingly, the irregular basicity order of amines in water (Me2NH>MeNH2>Me3N>NH3, as shown by the pKa values for the corresponding conjugated acids: 10.77>10.62>9.80>9.,246) (Dean, 1999) está contaminado por efectos solventes como se ilustra por el orden de basicidad sistemática de las aminas en fase gaseosa (Me3N>Me2NH>MeNH2>NH3) (Brauman, Riveros, & Blair, 1971)., Aunque el orden de basicidad de la fase gaseosa puede atribuirse al generalmente supuesto efecto alquilo +I (Carter, 2007), se ha observado una disminución de la densidad electrónica de nitrógeno a través de la sustitución de metilo por medio de cálculos de potencial electrostático Molecular (Baeten, de Proft, & Geerlings, 1995), lo que indica el comportamiento a –i para el grupo metilo., En realidad, el orden de basicidad de fase gaseosa de las aminas alifáticas debe atribuirse a la estabilización creciente de iones de amonio sustituidos debido a la polarizabilidad del grupo alquilo (Aue, Webb, & Bowers, 1976).
Las acididades relativas de los alcoholes en solución acuosa (H2O>MeOH>EtOH>iPrOH>tBuOH) también tienen se ha atribuido en algunos libros de texto al supuesto efecto alquilo +I (Johnson, 1999; Solomons, fryhle, & Snyder, 2016)., Dado que el orden de acidez inverso se encuentra en la fase gaseosa, las acididades relativas de los alcoholes en el agua deben atribuirse a las magnitudes más bajas de las entalpías de solvatación para aniones alcóxidos más grandes (Brauman & Blair, 1969).
la discusión sobre las propiedades electrónicas del grupo alquilo también se puede aplicar a los carbaniones. Por lo tanto, el orden de estabilidad de ‘libro de texto’ para carbaniones simples (metil>etil>isopropilo>terc-butilo) se ha atribuido al supuesto efecto inductivo +I de los grupos alquilos (Burrows et al.,, 2013; Chaloner, 2015; Roos & Roos, 2014; Smith, 2013). Sin embargo, se encuentra un orden irregular para las estabilidades de carbanión en fase gaseosa (TBU>Me>iPr>Et), de acuerdo con la concurrencia de dos efectos alquilos opuestos (DePuy et al., 1989): un mecanismo estabilizador a través de la polarizabilidad alquilo (es decir, n→σ* hiperconjugación) y una tendencia desestabilizadora (consistente con el papel de A +R, asumiendo un comportamiento similar a p para el par solitario de carbono).,
la estabilidad de otros intermedios de reacción también puede evaluarse sobre la base de los efectos del grupo alquilo. Por lo tanto, el conocido orden de estabilidad para carbocaciones (terciario>secundario>primario>metilo) se ha atribuido a veces a un efecto inductivo positivo (Chaloner, 2015; Roos & Foote, 2013; Burrows et al., 2013) aunque la escritura ambigua habitual impide determinar si ambas explicaciones corresponden a dos descripciones diferentes del mismo fenómeno o a dos mecanismos concurrentes que juegan en la misma dirección., De todos modos, el orden de estabilidad de las carbocaciones debe atribuirse a la hiperconjugación (por lo tanto, el comportamiento a+R en un orbital P vacante, el sistema π más simple), aunque otras interacciones (como la polarizabilidad alquilo) también están involucradas (Aue, 2011).
los radicales libres muestran el mismo orden de estabilidad que las carbocaciones, lo que indica la estabilización a través de la sustitución de alquilos. Aunque tal orden de estabilidad puede justificarse sobre la base de un comportamiento +I asumido, el efecto + R puede considerarse alternativamente, de manera análoga a la estabilización de radicales libres por átomos que contienen pares solitarios (Zipse, 2006).,
reactividad en solución
acididades relativas de ácidos carboxílicos simples en solución acuosa (ácido acético>ácido propiónico>ácido butírico) se han utilizado en algunos libros de texto para ilustrar el supuesto efecto +I del grupo alquilo (Sorrell, 2006). Curiosamente, el orden inverso se encuentra cuando se consideran entalpías (Christensen, Izatt, & Hansen, 1967), lo que indica que el orden de acidez en solución acuosa debe atribuirse a las entropías de hidratación., Por lo tanto, el orden de celosía significativo del agua líquida (entropía de vaporización equivalente a 118.89 Jmol–1K–1, en contraste con los valores típicos de ca. 88Jmol-1K-1 para la mayoría de los líquidos, Dean, 1999) puede introducir cambios considerables en la energética de la reacción. En particular, la hidratación de las moléculas apolares (o mitades) conduce a un nuevo ordenamiento de la red solvente (Blokzijl & Engberts, 1993). Como consecuencia, los efectos inductivos del grupo alquilo a partir de datos experimentales en solución acuosa a menudo están enmascarados por entropías de hidratación (Calder & Barton, 1971)., Acididades relativas de ácidos carboxílicos simples en fase gaseosa (Yamdagni & Kebarle, 1973) y acetonitrilo (Eckert et al., 2009) son consistentes con el papel principal desempeñado por las entropías de hidratación.
la menor acidez del ácido pivalico en comparación con el ácido acético, generalmente atribuida al supuesto efecto +I del grupo alquilo (Smith, 2008), se invierte cuando se consideran entalpías de reacción (Eckert et al., 2009).,
el supuesto efecto del grupo alquilo +I sobre la acidez de ácidos carboxílicos simples en solución acuosa puede atribuirse a un artefacto derivado de efectos solventes. Mientras que un aumento de volumen de solutos neutros conduce a un aumento de la entropía de hidratación, la relación inversa se encuentra para las especies iónicas (Graziano, 2009). Como consecuencia, la sustitución alquilo (a través de un aumento del volumen molecular) conduce a la estabilización (en términos de energía libre de Gibbs) del ácido no ionizado en el agua, así como la desestabilización del anión carboxilato correspondiente, lo que resulta en una disminución de la acidez.,
la mayor acidez del ácido fórmico en comparación con el ácido acético en solución acuosa (valores de pKa: 3.751 y 4.756, respectivamente, Dean, 1999) también se ha discutido en muchos libros de texto como un ejemplo de la aplicación de efectos inductivos (Hart, Hadad, Craine, & Hart, 2012; Hornback, 2006; Okuyama & maskill, 2014; Roos & Roos, 2014). Dado que las entalpías de reacción muy similares están involucradas en las reacciones de disociación de los ácidos fórmico y acético (Christensen et al.,, 1967), la mayor acidez del ácido fórmico debe atribuirse de hecho a las diferencias de entropía de hidratación.
conclusiones
una comprensión clara de los efectos inductivos y de resonancia es una clave importante para un aprendizaje sólido de la Química Orgánica (Mullins, 2008). Sorprendentemente, el grupo alquilo casi omnipresente se ha presentado incorrectamente en muchos libros de texto como un grupo σ-donante (+I). Sin embargo, un comportamiento dual es mostrado por los sustituyentes alquilos dependiendo de la hibridación del átomo vecino., Por lo tanto, los grupos alquilos Unidos a cadenas alifáticas se comportan como aceptores σ (- i, consistente con la electronegatividad más grande del carbono en relación con el hidrógeno), mientras que los unidos a los sistemas π actúan como donantes π (+R, Debido a interacciones hiperconjugativas). Una serie de datos experimentales y teóricos (momentos dipolares, RMN, ir y espectros UV, reactividad) concuerdan con este comportamiento dual.,
el análisis completo de todos los datos considerados aquí permite inferir un efecto pequeño-I, así como un comportamiento significativo +R para el grupo alquilo como una característica válida en todas las discusiones sobre las propiedades espectroscópicas y de reactividad de los compuestos orgánicos.
conflicto de intereses
el autor declara no tener conflicto de intereses.