Bowtie indexe le génome de référence en utilisant un schéma basé sur la transformation de Burrows-Wheeler (BWT) et L’index FM . Un index Bowtie pour le génome humain tient dans 2,2 Go sur disque et a une empreinte mémoire de aussi peu que 1,3 Go au moment de l’alignement, ce qui lui permet d’être interrogé sur un poste de travail avec moins de 2 Go de RAM.,
la méthode courante pour rechercher dans un index FM est l’algorithme de correspondance exacte de Ferragina et Manzini . Bowtie n’adopte pas simplement cet algorithme car la correspondance exacte ne permet pas d’erreurs de séquençage ou de variations génétiques. Nous introduisons deux nouvelles extensions qui rendent la technique applicable à l’alignement de lecture courte: un algorithme de retour en arrière conscient de la qualité qui permet les inadéquations et favorise les alignements de haute qualité; et « double indexation », une stratégie pour éviter un retour en arrière excessif., L’aligneur de Noeud papillon suit une politique similaire à celle de Maq, en ce sens qu’il permet un petit nombre de discordances dans la fin de haute qualité de chaque lecture, et il place une limite supérieure sur la somme des valeurs de qualité aux positions d’alignement incompatibles.
indexation Burrows-Wheeler
Le BWT est une permutation réversible des caractères d’un texte. Bien que développée à l’origine dans le contexte de la compression de données, l’indexation basée sur BWT permet de rechercher efficacement de gros textes dans une faible empreinte mémoire., Il a été appliqué aux applications bioinformatiques, y compris le comptage des oligomères , l’alignement du génome entier , la conception de la sonde de microréseau de carrelage et L’alignement Smith-Waterman sur une référence à taille humaine .
la transformation de Burrows-Wheeler d’un texte T, BWT(T), est construite comme suit. Le caractère $ est ajouté à T, où $ n’est pas dans T et est lexicographiquement inférieur à tous les caractères de T. La matrice de Burrows-Wheeler de T est construite comme la matrice dont les lignes comprennent toutes les rotations cycliques de T$. Les lignes sont ensuite triées lexicographiquement., BWT (T) est la séquence de caractères dans la colonne la plus à droite de la matrice de Burrows-Wheeler (Figure 1a). BWT(T) a la même longueur que le texte original T.
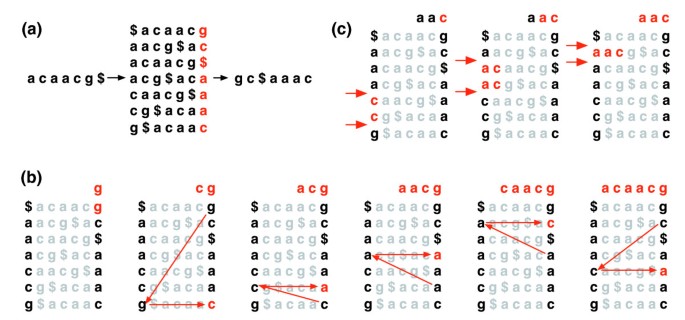
de Burrows-Wheeler transformer. a) la matrice et la transformation de Burrows-Wheeler pour « acaacg ». B) mesures prises par EXACTMATCH pour identifier la plage de lignes, et donc l’ensemble des suffixes de référence, préfixés par « aac »., (c) UNPERMUTE applique à plusieurs reprises le mappage last first (LF) pour récupérer le texte original (en rouge sur la ligne supérieure) à partir de la transformation de Burrows-Wheeler (en noir dans la colonne la plus à droite).
Cette matrice a une propriété appelée ‘le dernier premier (LF) cartographie ». La I occurrence du caractère X dans la dernière colonne correspond au même caractère de texte que la I occurrence de X dans la première colonne. Cette propriété sous-tend les algorithmes qui utilisent l’index BWT pour naviguer ou rechercher le texte., La Figure 1b illustre UNPERMUTE, un algorithme qui applique le mappage LF à plusieurs reprises pour recréer T à partir de BWT(T).
le mappage LF est également utilisé dans la correspondance exacte. Étant donné que la matrice est triée lexicographiquement, les lignes commençant par une séquence donnée apparaissent consécutivement. En une série d’étapes, L’algorithme EXACTMATCH (Figure 1c) calcule la plage de lignes matricielles commençant par des suffixes successivement plus longs de la requête. À chaque étape, la taille de la plage diminue ou reste la même., Lorsque l’algorithme est terminé, les lignes commençant par S0 (la requête entière) correspondent aux occurrences exactes de la requête dans le texte. Si la plage est vide, le texte ne contient pas la requête. IMPERMUTE est attribuable à Burrows et Wheeler et EXACTMATCH à Ferragina et Manzini . Voir le fichier de données supplémentaires 1 (Discussion supplémentaire 1) pour plus de détails.
rechercher des alignements inexacts
EXACTMATCH est insuffisant pour un alignement de lecture court car les alignements peuvent contenir des incohérences, qui peuvent être dues à des erreurs de séquençage, à des différences réelles entre les organismes de référence et de requête, ou les deux., Nous introduisons un algorithme d’alignement qui effectue une recherche de retour en arrière pour trouver rapidement des alignements qui satisfont une stratégie d’alignement spécifiée. Chaque caractère d’une lecture a une valeur de qualité numérique, avec des valeurs inférieures indiquant une probabilité plus élevée d’une erreur de séquençage. Notre Politique d’alignement permet un nombre limité de discordances et préfère les alignements où la somme des valeurs de qualité à toutes les positions incompatibles est faible.
la recherche se déroule de la même manière que EXACTMATCH, en calculant des plages de matrice pour des suffixes de requête successivement plus longs., Si la plage devient vide (aucun suffixe n’apparaît dans le texte), l’algorithme peut sélectionner une position de requête déjà appariée et y substituer une base différente, introduisant une non-concordance dans l’alignement. La recherche EXACTMATCH reprend juste après la position substituée. L’algorithme sélectionne uniquement les substitutions qui sont compatibles avec la stratégie d’alignement et qui produisent un suffixe modifié qui apparaît au moins une fois dans le texte. S’il existe plusieurs positions de substitution candidates, l’algorithme sélectionne goulûment une position avec une valeur de qualité minimale.,
Les scénarios de retour en arrière se déroulent dans le contexte d’une structure de pile qui se développe lorsqu’une nouvelle substitution est introduite et se rétrécit lorsque l’aligneur rejette tous les alignements candidats pour les substitutions actuellement sur la pile. Voir la Figure 2 pour une illustration de la façon dont la recherche peut se dérouler.
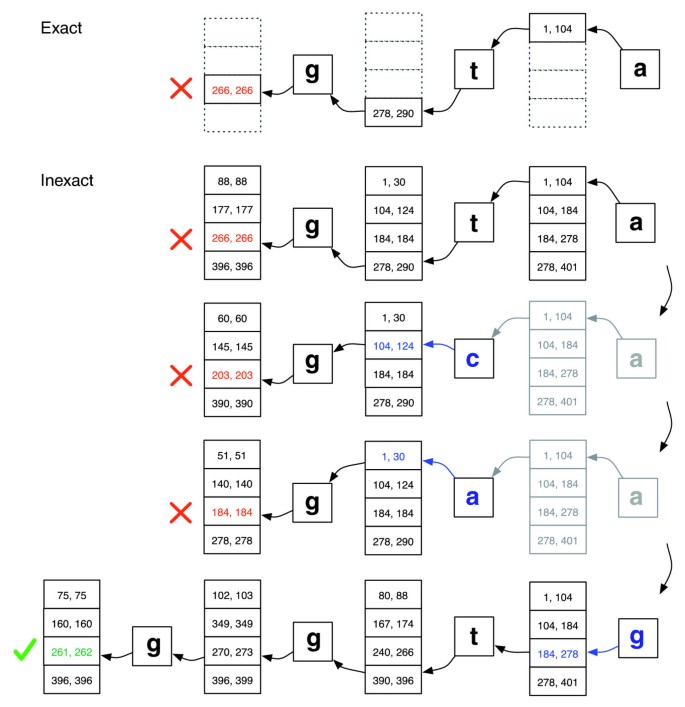
l’appariement Exact contre inexact d’alignement., Illustration de la façon dont EXACTMATCH (en haut) et l’aligneur de Bowtie (en bas) se déroulent lorsqu’il n’y a pas de correspondance exacte pour la requête ‘ggta’ mais qu’il y a un alignement d’une non-correspondance lorsque ‘a’ est remplacé par ‘g’. Les paires de nombres en boîte désignent des plages de lignes de matrice commençant par le suffixe observé jusqu’à ce point. Un X rouge marque l’endroit où l’algorithme rencontre une plage vide et abandonne (comme dans EXACTMATCH) ou fait marche arrière (comme dans l’algorithme inexact). Une coche verte indique que l’algorithme trouve une plage non vide délimitant une ou plusieurs occurrences d’un alignement à signaler pour la requête.,
En bref, Bowtie effectue une recherche qualitative, gourmande, randomisée et approfondie dans l’espace des alignements possibles. Si un alignement valide existe, alors Bowtie le trouvera (sous réserve du plafond backtrack discuté dans la section suivante). Parce que la recherche est gourmande, le premier alignement valide rencontré par Bowtie ne sera pas nécessairement le « meilleur » en termes de nombre de discordances ou en termes de qualité., L’utilisateur peut demander à Bowtie de continuer la recherche jusqu’à ce qu’il puisse prouver que tout alignement qu’il signale est « meilleur » en termes de nombre de discordances (en utilisant l’option best best). Dans notre expérience, ce mode est deux à trois fois plus lent que le mode par défaut. Nous prévoyons que le mode par défaut plus rapide sera préféré pour les grands projets de re-séquençage.
L’utilisateur peut également opter pour Bowtie pour signaler tous les alignements jusqu’à un nombre spécifié (option-k) ou tous les alignements sans limite sur le nombre (option-a) pour une lecture donnée., Si, au cours de sa recherche, Bowtie trouve N alignements possibles pour un ensemble donné de substitutions, mais que L’utilisateur n’a demandé que K alignements où K < N, Bowtie rapportera K Des n alignements sélectionnés au hasard. Notez que ces modes peuvent être beaucoup plus lents que la valeur par défaut. Dans notre expérience, par exemple,- k 1 est plus de deux fois plus rapide que-k 2.
retour arrière excessif
L’aligneur tel que décrit jusqu’à présent peut, dans certains cas, rencontrer des séquences qui provoquent un retour arrière excessif., Cela se produit lorsque l’aligneur passe la plupart de ses efforts à revenir en arrière sans succès vers des positions proches de la fin 3′ de la requête. Bowtie atténue le retour en arrière excessif avec la nouvelle technique de « double indexation ». Deux indices du génome sont créés: l’un contenant le BWT du génome, appelé index « forward », et un second contenant le BWT du génome avec sa séquence de caractères inversée (non complétée par inversion) appelé index « mirror ». Pour voir comment cela aide, envisagez une stratégie de correspondance qui autorise une non-concordance dans l’alignement., Un alignement valide avec une non-concordance tombe dans l’un des deux cas selon lesquels la moitié de la lecture contient la non-concordance. Bowtie procède en deux phases correspondant à ces deux cas. La Phase 1 charge l’index direct en mémoire et appelle l’aligneur avec la contrainte qu’il ne peut pas remplacer aux positions dans la moitié droite de la requête. La Phase 2 utilise l’index miroir et appelle l’aligneur sur la requête inversée, avec la contrainte que l’aligneur ne peut pas remplacer aux positions dans la moitié droite de la requête inversée (la moitié gauche de la requête d’origine)., Les contraintes de retour en arrière dans la moitié droite empêchent un retour en arrière excessif, alors que l’utilisation de deux phases et de deux indices maintient une sensibilité totale.
malheureusement, il n’est pas possible d’éviter complètement un retour en arrière excessif lorsque les alignements sont autorisés à avoir deux ou plusieurs incompatibilités. Dans nos expériences, nous avons observé qu’un retour en arrière excessif n’est significatif que lorsqu’une lecture a de nombreuses positions de mauvaise qualité et ne s’aligne pas ou s’aligne mal à la référence., Ces cas peuvent déclencher plus de 200 retours en arrière par lecture, car il existe de nombreuses combinaisons juridiques de positions de mauvaise qualité à explorer avant que toutes les possibilités ne soient épuisées. Nous réduisons ce coût en imposant une limite sur le nombre de retours en arrière autorisés avant la fin d’une recherche (par défaut: 125). La limite empêche la déclaration de certains alignements légitimes de mauvaise qualité, mais nous nous attendons à ce qu’il s’agisse d’un compromis souhaitable pour la plupart des applications.,
Phased MAQ-like search
Bowtie permet à l’utilisateur de sélectionner le nombre de discordances autorisées (par défaut: deux) dans la fin de haute qualité d’une lecture (par défaut: les 28 premières bases) ainsi que la distance de qualité maximale acceptable de l’alignement global (par défaut: 70). Les valeurs de qualité sont supposées suivre la définition de PHRED, où p est la probabilité d’erreur et Q = -10log p.
le complément lu et son complément inverse sont candidats à l’alignement sur la référence. Pour plus de clarté, cette discussion ne tient compte que de l’orientation prospective., Voir le fichier de données supplémentaires 1 (Discussion supplémentaire 2) pour une explication de la façon dont le complément inverse est incorporé.
Les 28 premières bases sur la fin de haute qualité de la lecture sont appelées la « graine ». La graine se compose de deux moitiés: la 14 pb à l’extrémité de haute qualité (généralement l’extrémité 5′) et la 14 pb à l’extrémité de faible qualité, appelées respectivement la ‘hi-half’ et la ‘lo-half’., En supposant la politique par défaut (deux incompatibilités permises dans la semence), un alignement déclarable se classera dans l’un des quatre cas suivants: Pas de non-concordance dans la semence (cas 1); Pas de non-concordance dans hi-half, une ou deux non-concordance dans lo-half (cas 2); pas de non-concordance dans lo-half, une ou deux non-concordance dans hi-half (cas 3); et une non-concordance dans hi-half, une non-concordance dans lo-half (cas 4).
Tous les cas autorisent n’importe quel nombre de discordances dans la partie non nécessaire de la lecture et tous les cas sont également soumis à la contrainte de distance de qualité.,
L’algorithme du nœud papillon se compose de trois phases qui alternent entre l’utilisation des indices avant et miroir, comme illustré à la Figure 3. La Phase 1 utilise l’index miroir et appelle l’aligneur pour trouver des alignements pour les cas 1 et 2. Les Phases 2 et 3 coopèrent pour trouver des alignements pour le cas 3: la Phase 2 trouve des alignements partiels avec des inadéquations uniquement dans la moitié hi et la phase 3 tente d’étendre ces alignements partiels en alignements complets. Enfin, la phase 3 appelle l’aligneur pour trouver des alignements pour le cas 4.,

Les trois phases du Papillon algorithme pour la Maq-comme la politique. Une approche triphasée permet de trouver des alignements pour les deux cas de non-concordance 1 à 4 tout en minimisant le retour en arrière. La Phase 1 utilise l’index miroir et appelle l’aligneur pour trouver des alignements pour les cas 1 et 2. Les Phases 2 et 3 coopèrent pour trouver des alignements pour le cas 3: la Phase 2 trouve des alignements partiels avec des discordances uniquement dans la moitié hi, et la phase 3 tente d’étendre ces alignements partiels en alignements complets., Enfin, la phase 3 appelle l’aligneur pour trouver des alignements pour le cas 4.
résultats de Performance
Nous avons évalué la performance de Bowtie en utilisant les lectures du projet pilote 1,000 Genomes (National Center for Biotechnology Information Short Read Archive:SRR001115). Au total, 8,84 millions de lectures, soit environ une voie de données provenant d’un instrument Illumina, ont été réduites à 35 PB et alignées sur le génome humain de référence . Sauf indication contraire, les données lues ne sont pas filtrées ou modifiées (en plus du rognage) de la façon dont elles apparaissent dans l’archive., Cela conduit à environ 70% à 75% des lectures s’alignant quelque part sur le génome. D’après notre expérience, cela est typique pour les données brutes de l’archive. Un filtrage plus agressif entraîne des taux d’alignement plus élevés et un alignement plus rapide.
Toutes les exécutions ont été effectuées sur un seul processeur. Les accélérations de nœud papillon ont été calculées comme un rapport des temps d’alignement de l’horloge murale. Les temps d’horloge murale et de CPU sont donnés pour démontrer que la charge d’entrée/sortie et la contention de CPU ne sont pas des facteurs significatifs.
le temps nécessaire pour construire L’index du nœud papillon n’était pas inclus dans les temps de fonctionnement du nœud papillon., Contrairement aux outils concurrents, Bowtie peut réutiliser un index pré-calculé pour le génome de référence sur de nombreuses pistes d’alignement. Nous prévoyons que la plupart des utilisateurs téléchargeront simplement de tels indices à partir d’un référentiel public. Le site Bowtie fournit des indices pour les constructions actuelles des génomes humain, chimpanzé, souris, Chien, rat et Arabidopsis thaliana, ainsi que de nombreux autres.
Les résultats ont été obtenus sur deux plates-formes matérielles: une station de travail de bureau avec un processeur Intel Core 2 de 2,4 GHz et 2 Go de RAM; et un serveur de grande mémoire avec un processeur AMD Opteron à quatre cœurs de 2,4 GHz et 32 Go de RAM., Ceux-ci sont notés » PC » et « serveur », respectivement. Le PC et le serveur exécutent Red Hat Enterprise Linux en tant que version 4.
comparaison avec SOAP et Maq
Maq est un aligneur populaire qui fait partie des outils open source concurrents les plus rapides pour aligner des millions de lectures Illumina sur le génome humain. SOAP est un autre outil open source qui a été signalé et utilisé dans des projets à lecture courte .
le tableau 1 présente les performances et la sensibilité de Bowtie v0.9.6, SOAP V1.10 et Maq v0.6.6. SOAP n’a pas pu être exécuté sur le PC car L’empreinte mémoire de SOAP dépasse la mémoire physique du PC. Le » savon.,la version contig du binaire SOAP a été utilisée. À titre de comparaison avec SOAP, Bowtie a été invoqué avec ‘ – v 2 ‘pour imiter la Politique de correspondance par défaut de SOAP (qui permet jusqu’à deux incompatibilités dans l’alignement et ne tient pas compte des valeurs de qualité), et avec’ max maxns 5 ‘ pour simuler la politique par défaut de SOAP consistant à filtrer les lectures avec cinq bases de Pour la comparaison Maq, Bowtie est exécuté avec sa stratégie par défaut, qui imite la stratégie par défaut de Maq consistant à autoriser jusqu’à deux incompatibilités dans les 28 premières bases et à appliquer une limite globale de 70 sur la somme des valeurs de qualité à toutes les positions de lecture incompatibles., Pour rendre L’empreinte mémoire de Bowtie plus comparable à celle de Maq, Bowtie est invoqué avec l’option’- z ‘ dans toutes les expériences pour s’assurer que seul l’index avant ou miroir réside en mémoire en même temps.
le nombre de lectures alignées indique que SOAP (67,3%) et Bowtie-v 2 (67,4%) ont une sensibilité comparable. Parmi les lectures alignées par SOAP ou Bowtie, 99,7% étaient alignées par les deux, 0,2% étaient alignées par Bowtie mais pas SOAP, et 0,1% étaient alignées par SOAP mais pas Bowtie. Maq (74.7%) et Bowtie (71.9%) ont également une sensibilité à peu près comparable, bien que Bowtie accuse un retard de 2.8%., Parmi les lectures alignées par Maq ou Bowtie, 96,0% étaient alignées par les deux, 0,1% étaient alignées par Bowtie mais pas Maq, et 3,9% étaient alignées par Maq mais pas Bowtie. Parmi les lectures mappées par Maq mais pas Bowtie, presque toutes sont dues à une flexibilité dans l’algorithme d’alignement de Maq qui permet à certains alignements d’avoir trois incompatibilités dans la graine. Le reste des lectures cartographiées par Maq mais pas Bowtie sont dues au plafond de retour en arrière de Bowtie.
la documentation de Maq mentionne que les lectures contenant des « artefacts poly-A » peuvent nuire aux performances de Maq., Le tableau 2 présente les performances et la sensibilité de Bowtie et Maq lorsque l’ensemble de lecture est filtré à l’aide de la commande ‘catfilter’ de Maq pour éliminer les artefacts poly-A. Le filtre élimine 438 145 lectures sur 8 839 010. D’autres paramètres expérimentaux sont identiques à ceux des expériences du tableau 1, et les mêmes observations sur la sensibilité relative de Bowtie et Maq s’appliquent ici.
longueur et performances de lecture
à mesure que la technologie de séquençage s’améliore, les longueurs de lecture dépassent les 30 à 50 bp couramment observées dans les bases de données publiques d’aujourd’hui., Bowtie, Maq et SOAP prennent en charge les lectures de longueurs jusqu’à 1 024, 63 et 60 bp, respectivement, et les versions maq 0.7.0 et ultérieures prennent en charge les longueurs de lecture jusqu’à 127 bp. Le tableau 3 montre les résultats de performance lorsque les trois outils sont chacun utilisés pour aligner trois ensembles de lectures non découpées de 2 M, un ensemble de 36 PB, un ensemble de 50 PB et un ensemble de 76 PB, sur le génome humain sur la plate-forme serveur. Chaque ensemble de 2 M est échantillonné aléatoirement à partir d’un ensemble plus grand (NCBI Short Read Archive: SRR003084 pour 36-bp, SRR003092 pour 50-bp, SRR003196 pour 76-bp)., Les lectures ont été échantillonnées de telle sorte que les trois ensembles de 2 M ont un taux d’erreur uniforme par base, calculé à partir des qualités Phred par base. Toutes les lectures passent par le « catfilter » de Maq.
Bowtie est exécuté à la fois dans son mode par défaut de type Maq et dans son mode ‘-v 2’ de type SOAP. Bowtie reçoit également l’option ‘- z ‘ pour s’assurer que seul l’index avant ou miroir réside en mémoire en même temps. Maq v0.7.1 a été utilisé à la place de Maq v0.6.6 pour l’ensemble de 76 bp Car v0. 6.,6 ne peut pas aligner les lectures supérieures à 63 bp. SOAP n’a pas été exécuté sur l’ensemble 76 bp car il ne prend pas en charge les lectures supérieures à 60 bp.
les résultats montrent que l’algorithme de Maq évolue mieux globalement vers des longueurs de lecture plus longues que Bowtie ou SOAP. Cependant, le nœud papillon en mode « -v 2 » ressemblant à du savon évolue également très bien. Bowtie dans son mode de type MAQ par défaut évolue bien de 36 bp à 50 bp lit, mais est sensiblement plus lent pour 76 bp lit, bien qu’il soit encore plus d’un ordre de grandeur plus rapide que Maq.,
performances parallèles
L’alignement peut être parallélisé en répartissant les lectures entre les threads de recherche simultanés. Papillon permet à l’utilisateur de spécifier un nombre de threads (option -p); noeud Papillon lance ensuite le nombre de threads à l’aide de la bibliothèque pthreads. Les threads Bowtie se synchronisent les uns avec les autres lors de la récupération des lectures, de la sortie des résultats, de la commutation entre les index et de l’exécution de diverses formes de comptabilité globale, telles que le marquage d’une lecture comme « terminée »., Sinon, les threads sont libres de fonctionner en parallèle, ce qui accélère considérablement l’alignement sur les ordinateurs avec plusieurs cœurs de processeur. L’image mémoire de l’index est partagée par tous les threads, de sorte que l’empreinte n’augmente pas substantiellement lorsque plusieurs threads sont utilisés. Le tableau 4 montre les résultats de performance pour l’exécution de Bowtie v0.9. 6 sur le serveur à quatre cœurs avec un, deux et quatre threads.,
construction D’Index
Bowtie utilise un algorithme d’indexation flexible qui peut être configuré pour faire un compromis entre l’utilisation de la mémoire et le temps d’exécution. Le tableau 5 illustre ce compromis lors de l’indexation de l’ensemble du génome humain de référence (NCBI build 36.3, contigs). Les exécutions ont été effectuées sur la plate-forme serveur. L’indexeur a été exécuté quatre fois avec des limites supérieures différentes sur l’utilisation de la mémoire.,
les temps rapportés se comparent favorablement aux temps d’alignement des outils concurrents qui effectuent l’indexation pendant l’alignement. Moins de 5 heures sont nécessaires pour que Bowtie construise et interroge un index humain entier avec 8,84 millions de lectures du projet 1,000 Genome (NCBI Short Read Archive:SRR001115) sur un serveur, plus de six fois plus rapide que l’exécution équivalente de Maq., La ligne la plus basse illustre que L’indexeur Bowtie, avec des arguments appropriés, est suffisamment économe en mémoire pour fonctionner sur un poste de travail typique avec 2 Go de RAM. Fichier de données supplémentaires 1 (Complémentaire des discussions 3 et 4) explique l’algorithme et le contenu de l’indice.
logiciel
Bowtie est écrit en C++ et utilise la bibliothèque SeqAn . Le convertisseur vers le format de mappage Maq utilise le code de Maq.