Einleitung
Substituent Effekte bilden ein Schlüsselkonzept für das Verständnis von Reaktivität und spektroskopischem Verhalten organischer Verbindungen (Krygowski & St??pień, 2005). In einem einfachen Ansatz können Substitutionseffekte nach dem Mechanismus der Wechselwirkung mit dem reaktiven Zentrum als induktive (durch σ-Bindungen) oder Resonanzeffekte (durch π-Bindungen) klassifiziert werden., Nichtsdestoweniger wären einige weitere Begriffe (wie sterische, Feld-oder Lösungsmitteleffekte) für eine gründliche Beschreibung der Substituenteneffekte erforderlich.
Seit der Ingold ‚ s classification of electronic substituent effects (Ingold, 1953) wird die Alkylgruppe in den meisten Lehrbüchern für organische Chemie (Burrows, Holman, Parsons, Pilling, & Price, 2013; Hornback, 2006; Roos & Roos, 2014; Smith, 2013; Vollhardt & Schore, 2014)., Dennoch sollte die Kritik der Eğe an einem so simplen Standpunkt beachtet werden:
„In Wasser ist Propansäure etwas schwächer als Essigsäure. Die Art der induktiven Wirkung einer Alkylgruppe wird von Chemikern diskutiert. Alkylgruppen stabilisieren Carbokationen und scheinen in dieser Rolle elektronenfrei zu sein. Sie erhöhen auch die Basizität von Aminen, was wiederum darauf hindeutet, dass sie Elektronen freisetzen. Auf der anderen Seite ist zwar Tert-Butylalkohol (pKa 19) in Wasser eine schwächere Säure als Ethanol (pKa 17), in der Gasphase jedoch eine stärkere Säure., Diese experimentelle Beobachtung legt nahe, dass Alkylgruppen sowohl Anionen als auch Kationen stabilisieren können und dass die Solvation eine wichtige Rolle bei der Bestimmung relativer Aziditäten spielt. Daher ist ein Wort der Vorsicht notwendig. Die relativen Aziditäten, auf denen die in diesem Kapitel dargestellten Verallgemeinerungen beruhen, wurden in Wasser bestimmt. In der Gasphase sind häufig Umkehrungen in der Reihenfolge verwandter Verbindungen zu sehen.“(Eğe, 1999, S. 107)
Einige Alkylsubstitutionseffekte wurden in Lehrbüchern oft auf widersprüchliche oder rätselhafte Weise erklärt., So werden chemische Verschiebungsunterschiede zwischen CH3-und CH2-Gruppen im Hornback-Buch der Tatsache zugeschrieben, dass“ Kohlenstoff etwas elektronegativer ist als Wasserstoff “ (Hornback, 2006, S. 549), obwohl die Alkylgruppe zuvor als schwach induktiver elektronenspendender Substituent klassifiziert wurde (Hornback, 2006, S. 117). Im Lehrbuch des Vollhardt wird die Beziehung zwischen chemischen Verschiebungen der Methylgruppe für eine Reihe von CH3X-Verbindungen und der X-Elektronegativität durch eine Tabelle veranschaulicht, in der ein Eintrag für X=methyl fehlt (Vollhardt & Schore, 2014, p., 389), wodurch das unbequeme Kohlenstoffproblem vermieden wird.
Ich zeige hier, dass sich die Alkylgruppe als a –I+R Substituent verhält. Obwohl einige Faktoren (wie Feld -, sterische oder Lösungsmitteleffekte) bei diesem Ansatz implizit ignoriert werden, können viele derzeit verfügbare theoretische und experimentelle Beweise auf einfache Weise beschrieben werden.
Eine Cδ–Hδ+ – Bindungspolarisation wurde experimentell für Methan beobachtet (Lazzeretti, Zanasi, & Raynes, 1987), konsistent mit der größeren Elektronegativität von Kohlenstoff relativ zu Wasserstoff, 2.55 vs. 2.,20 in der Pauling-Skala (Allred, 1961). Ein solches Polarisationsmuster ermöglicht die Vorhersage der Dipolmomentrichtung einfacher Kohlenwasserstoffe durch additive Modelle, obwohl die quantitative Übereinstimmung normalerweise bescheiden ist (2-Methylpropan: 0,3 D geschätzt vs. 0,132 D experimentell) (Dean, 1999).
Da Wasserstoff als Standard in der Ingold-Klassifikation von Substituenten verwendet wird (Krygowski & St??pień, 2005), sollte die Alkylgruppe als a –I-Substituent klassifiziert werden (daher eine σ-Elektron-entnehmende Gruppe). Eine solche Rolle ist in Abb., 1 für die σ-Bindungspolarisation von einem beliebigen Atom Y zu einer Methylgruppe, obwohl die umgekehrte Bindungspolarisation erwartet wird, wenn Y elektronegativer ist als Kohlenstoff (z. B. Chlor).
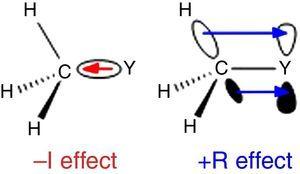
– I (links) und +R(rechts) Effekte einer an ein Atom gebundenen Methylgruppe Y.
Für Alkylgruppen wird ein anderes Verhalten gefunden, wenn sie an sp2-oder sp–hybridisierte Atome aufgrund der Elektronendichte gebunden sind, die von Alkyl–C-H-oder C-C σ-Bindungen an das leere p-Orbital des zusammenhängenden Atoms (das einfachste π-System), wie in Abb. 1. Somit kann die Abnahme der Gasphasensäure für Phenol und Benzoesäure durch p-Methylsubstitution (McMahon & Kebarle, 1977) nur auf einen signifikanten π-Donor-Effekt für Methylsubstituenten zurückgeführt werden (tatsächlich größer als der für Methoxygruppe)., Die Alkylgruppe sollte jedoch aufgrund des Fehlens einsamer Elektronenpaare als atypischer π-Donor-Substituent betrachtet werden. Eine solche σ-Bindung / π-System-Wechselwirkung, die als Hyperkonjugation bezeichnet wird (Mullins, 2012), kann leicht in Analogie zum π-Donor-Verhalten eines einsamen paartragenden Atoms (z. B. Chlor) zu einem leeren p–Orbital erklärt werden, obwohl C–C-oder C-H-Bindungen (anstelle von Elektronenlonenpaaren) der Alkylgruppe als elektronenfreisetzende Einheiten an hyperkonjugativen Wechselwirkungen beteiligt sind., Interessanterweise sind π→σ * – Wechselwirkungen (negative Hyperkonjugation) für Alkylgruppen, denen elektronegative Atome fehlen, normalerweise vernachlässigbar (Bocca, Pontes, & Basso, 2004).
Einige molekulare Strukturmerkmale können anhand der Alkylgruppeneigenschaften rationalisiert werden. Zum Beispiel die größeren CO-Bindungslängen in Methylketonen (Aceton: exp. 1.210 Å, calc. 1.193 Å) im Vergleich zu den verwandten Aldehyden (Acetaldehyd: exp. 1.209 Å, calc. 1.,188Å) (Berry, Waltman, Pacansky, & Hagler, 1995) kann der Stabilisierung der zwitterionischen Resonanzform zugeschrieben werden (siehe Abb. 2) durch Alkylgruppe π-Spende an das Carbonylcarbonatom, wodurch das Doppelbindungsmerkmal der Carbonylgruppe geschwächt wird.
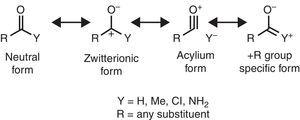
Neutrale (linke) und zwitterionische (rechte) Resonanzformen einer Carbonylverbindung.
Hyperkonjugative Wechselwirkungen sind abhängig von der Anordnung von C–H (oder C–C) – Bindungen relativ zum p-Orbital des zusammenhängenden Atoms Y, der effektivsten Wechselwirkung, die einer fast parallelen Anordnung entspricht. Zum Beispiel ist die Toluol-Csp3-H-Bindung fast senkrecht zur Rahmenebene etwas länger als die anderen Csp3-H-Bindungen (um 0,002 Å, Hameka & Jensen, 1996)., Die Geometrieabhängigkeit der Hyperkonjugation ermöglicht die Erklärung der Konformationsanalyse von methylsubstituierten ungesättigten Verbindungen wie Propen (Liberles, O ‚ Leary, Eilers, & Whitman, 1972) oder Acetaldehyd (Muñoz-Caro, Niño, & Moule, 1994).
Als bekannte Konsequenz des π-Donor-Verhaltens der Alkylgruppe ergibt die Alkylsubstitution elektronenreichere Alkene und Arene (Libit & Hoffmann, 1974)., Die hohe Reaktivität eines alkylsubstituierten Arens in einer SEAr-Reaktion kann somit auf die Stabilisierung des entsprechenden Wheland-Intermediates durch π-Elektronenspende zurückgeführt werden.
Das-I+R-Verhalten der Alkylgruppe ermöglicht es, eine Reihe von Merkmalen von alkylsubstituierten Verbindungen wie Dipolmomente, spektroskopische Eigenschaften und Reaktivität (in Gasphasen-und Lösungsmedien) zu erklären, wie unten gezeigt.
Dipolmomente
Das Elektronenrückzugsverhalten der Alkylgruppe in aliphatischen Verbindungen spiegelt sich auch in Dipolmomenten wider., Somit sind die Dipolmomentvektoren für Propan und 2-Methylpropan (Tasi et al., 1997) sowie einige substituierte Bicyclooctane (Böhm & Exner, 2004) auf den Rückzugseffekt (–I) der Methylgruppe im Vergleich zu Wasserstoff zurückgeführt werden (siehe Abb. 3).
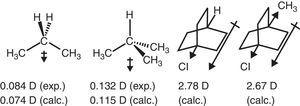
Dipolmomente von Propan und substituierten Bicyclooctanen.
Im Gegensatz dazu ist der π-Donor-Charakter der Methylgruppe (+R) erforderlich, um das Vorhandensein von Dipolmomenten von Nitrobenzen und Benzonitril durch p-Methylsubstitution (Brown, 1959) zu erklären (siehe Abb. 4).
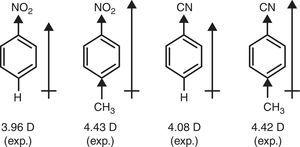
Dipolmomente von Benzolderivaten.
Molekulare Dipolmomente können mit aktuellen Rechenmethoden zuverlässig berechnet werden., Interessanterweise sind die berechneten Dipolmomentvektoren für einen Satz einfacher Kohlenwasserstoffe (Tasi et al., 1997) haben es ermöglicht, eine doppelte Rolle für die Methylgruppe abzuleiten: Elektronentzug bei Bindung an sp3-Kohlenstoffatome, aber Elektronenspende bei Bindung an sp2-oder sp3-Kohlenstoff.
Ein solches duales Verhalten des Alkylsubstituenten wird auch für Heteroatom-tragende Verbindungen beobachtet. Somit wird eine allmähliche Dipolmomentabnahme bei sukzessiver Methylsubstitution an Ammoniak beobachtet (NH3, 1,47 D; MeNH2, 1,31 D; Me2NH, 1,01 D; Me3N, 0.,61 D) (Le Fèvre & Russell, 1947), in Übereinstimmung mit der fortschreitenden Abnahme der Stickstoffelektronendichte (Hhre & Pople, 1970). Im Gegensatz dazu wird eine Dipolmomentverstärkung (von 1,53 D auf 1,68 D) (Nelson, Lide, & Maryott, 1967) für die Substitution von N, N-Dimethyl auf Anilin gefunden (Targema,Obi-Egbedi, & Adeoye, 2013), konsistent mit der Erhöhung des π-Spenderzeichens für die Aminogruppe (Hinchliffe & Kidd, 1980) aufgrund +R Beiträge von Methylsubstituenten.,
Spektroskopische Eigenschaften
Spektroskopische Eigenschaften vieler organischer Verbindungen können leicht rationalisiert werden, indem das Verhalten von a –I+R für die Alkylgruppe als allgemeines Merkmal angenommen wird. Somit kann die chemische NMR – Verschiebung eines Atoms als experimentelles Maß für die Elektronendichte an der entsprechenden Kernposition angesehen werden, obwohl auch andere Effekte – wie anisotrope Magnetfelder-beteiligt sein können. Downfield Verschiebungen induziert durch einen Methylsubstituenten auf sp3 Kohlenstoffatome (+9,6 ppm in 13C NMR) oder die entsprechenden gebundenen Wasserstoffatome (+0.,63ppm in 1H NMR) (Pretsch, Bühlmann, & Badertscher, 2009) mit dem Verhalten typischer –I-Gruppen (wie Halogenatome) übereinstimmen.
Alkylsubstitutionseffekte auf NMR Chemische Verschiebungen von Alkenen zeigen eine Abnahme der Elektronendichte in α-Position (+12,9 ppm für 13C NMR; +0,45 ppm für 1H NMR) sowie eine Dichteerhöhung in β –Position (-7,4 ppm für 13C; -0,31/-0,40 ppm für 1H), konsistent mit a-I+R-Effekt, obwohl auch anisotrope Effekte (wie Ringströme) eine Rolle spielen können. Ein solches a-I+R-Verhalten findet sich auch für Alkine, gemäß 13C NMR Spektroskopie (+8.,5ppm für α-position, -3.6 ppm für β-position).
Das dichotome Verhalten von Alkylsubstituenten auf π-Systemen (Elektronendichteerhöhung für α-Atom, Elektronendichteabnahme für β-Atom) kann nicht auf der Grundlage eines einfachen Verhaltens (wie a +I-Effekt) erklärt werden.
A-I+R-Verhalten (Meier, 2007) wird durch 15N-NMR – Spektroskopie zur Alkylsubstitution an Aminen und Amiden in Abhängigkeit von der Stickstoffhybridisierung beobachtet (Downfield-Verschiebungen für aliphatische Amine, Upfield-Verschiebungen für Nsp2-tragende Verbindungen-wie Aniline und Amide).,
NMR-Kopplungskonstanten sind ebenfalls abhängig von substituenten elektronischen Eigenschaften (sowie einigen geometrischen Merkmalen). Somit wird eine signifikante Abnahme für 1H–1H-Kopplungskonstanten durch Methylsubstitution (trans, -2,3 Hz; cis, -1,6 Hz; gem, -0,4 Hz) in qualitativer Übereinstimmung mit Daten typischer elektronenabziehender Gruppen wie dem Fluoratom (trans, -6,3 Hz; cis, -6,9 Hz; gem, -5,7 Hz) gefunden. Der positive Beitrag zur Methylsubstitution auf 13C-1H-Kopplungskonstanten aliphatischer Verbindungen (+1.,0Hz), ist auch qualitativ konsistent mit denen aus anderen-I Gruppen (Fluor, +24Hz).
Die Infrarotspektroskopie ist auch empfindlich gegenüber Substituenteneigenschaften, wie die Koeffizientenfrequenz von Carbonylverbindungen in Abhängigkeit vom entsprechenden Substituenten Y zeigt, der in Bezug auf Resonanzformen rationalisiert werden kann (Abb. 2). Durch Einnahme eines aliphatischen Aldehyds (ca., 1725cm-1) als Referenz kann die durch einen +I–Substituenten induzierte Rotverschiebung (Wellenzahlabnahme) (Acetyltrimethylsilan, 1645cm-1: Soderquist & Hsu, 1982) auf die Stabilisierung der zwitterionischen Form zurückgeführt werden. Stattdessen provozierte das Blueshift durch a-I-Substituenten (Acylchloride, >1800cm-1: Pretsch et al., 2009) kann durch zwei alternative oder gleichzeitige Mechanismen erklärt werden (Destabilisierung der zwitterionischen Form und/oder Beitrag einer acyliumionen tragenden Form). Schließlich provozierten die Rotschüsse durch +R Substituenten (Amide, ca., 1680cm–1: Pretsch et al., 2009) kann dem Beitrag einer bestimmten Resonanzform zugeschrieben werden. Die leichte Rotverschiebung induziert durch Alkylgruppe (Methylketone, ca. 1715cm-1) zeigt einen Nettoelektronenspendendeneffekt (daher eine Dominanz des +R-Effekts gegenüber – I-Eigenschaften). Der Nettospendereffekt der carbonylgebundenen Alkylgruppe stimmt mit dem größeren Dipolmoment von Aceton (2,88 D) relativ zu Formaldehyd (2,33 D) überein (Nelson et al., 1967).
Der Einfluss der Alkylgruppe auf UV–Vis-Spektren vieler Verbindungen lässt sich auch im Hinblick auf elektronische Effekte erklären., Somit stimmen bathochrome Verschiebungen,die durch Alkylgruppen auf UV-Absorptionsbanden von α, β-ungesättigten Verbindungen (+10 nm in α-Position, +12 nm in β-Position), konjugierten Polyenen (+5 nm) oder Benzolderivaten (+3,0 nm) induziert werden, qualitativ mit Wirkungen typischer π-Donorgruppen (z. B. Chlor) überein.
Gasphase Säure-Base-Reaktivität
Relative Basizitäten aliphatischer Amine in wässriger Lösung wurden der angenommenen +I-Wirkung der Alkylgruppe zugeschrieben (Sorrell, 2006)., Interestingly, the irregular basicity order of amines in water (Me2NH>MeNH2>Me3N>NH3, as shown by the pKa values for the corresponding conjugated acids: 10.77>10.62>9.80>9.,246) (Dean, 1999) kontaminiert ist, durch Lösungsmittel-Effekte wie dargestellt durch die systematische Reihenfolge der basizität von Aminen in der Gasphase (Me3N>Me2NH>MeNH2>NH3) (Brauman, Riveros, & Blair, 1971)., Obwohl die Gasphasenbasizitätsreihenfolge dem üblicherweise angenommenen +I –Alkyl-Effekt zugeschrieben werden kann (Carter, 2007), wurde mittels molekularer elektrostatischer Potentialberechnungen (Baeten, De Proft, & Geerlings, 1995) tatsächlich eine Abnahme der Stickstoffelektronendichte durch Methylsubstitution beobachtet, was auf ein a-I-Verhalten für die Methylgruppe hinweist., Tatsächlich sollte die Gasphasenbasizitätsreihenfolge aliphatischer Amine der zunehmenden Stabilisierung substituierter Ammoniumionen aufgrund der Alkylgruppenpolarisierbarkeit zugeschrieben werden (Aue, Webb, & Bowers, 1976).
Relative acidities der Alkohole in wässriger Lösung (H2O>MeOH>EtOH>iPrOH>tBuOH), wurden auch zurückzuführen, dass in einigen Lehrbüchern zu den vermeintlichen alkyl – +I-Effekt (Johnson, 1999; Solomons, Fryhle, & Snyder, 2016)., Da die umgekehrte Säurereihenfolge in der Gasphase gefunden wird, sollten die relativen Aziditäten von Alkoholen in Wasser den niedrigeren Größen der Solvationsenthalpien für größere Alkoxidanionen zugeschrieben werden (Brauman & Blair, 1969).
Die Diskussion über elektronische Eigenschaften von Alkylgruppen kann auch auf Carbanionen angewendet werden. Somit wurde die ‚Lehrbuch‘-Stabilitätsreihenfolge für einfache Carbanionen (methyl>ethyl>isopropyl>tert-Butyl) der angenommenen +I-induktiven Wirkung von Alkylgruppen zugeschrieben (Burrows et al.,, 2013; Chaloner, 2015; Roos & Roos, 2014; Smith, 2013). Es wird jedoch eine unregelmäßige Reihenfolge für Gasphasenkarbanionsstabilitäten gefunden (tBu>Me>iPr>Et), in Übereinstimmung mit der Gleichzeitigkeit zweier entgegengesetzter Alkyleffekte (DePuy et al., 1989): ein stabilisierender Mechanismus, durch alkyl-Polarisierbarkeit (, dass ist, n→σ* hyperconjugation) und eine destabilisierende Tendenz (durchweg mit a +R-Rolle, durch die Annahme einer p-wie Verhalten für die carbon lone pair).,
Die Stabilität anderer Reaktionszwischenprodukte kann auch anhand von Alkylgruppeneffekten beurteilt werden. Daher wurde die bekannte Stabilitätsreihenfolge für Carbokationen (tertiär>sekundär>primär>methyl) manchmal einem positiven induktiven Effekt zugeschrieben (Chaloner, 2015; Roos & Roos, 2014)., Interessanterweise wird Hyperkonjugation in vielen Lehrbüchern als alternative Erklärung für die Stabilitätsreihenfolge von Carbokationen dargestellt (Brown, Iverson, Anslyn, & Foote, 2013; Burrows et al., 2013) obwohl das übliche mehrdeutige Schreiben verhindert, dass festgestellt wird, ob beide Erklärungen entweder zwei verschiedenen Beschreibungen desselben Phänomens oder zwei gleichzeitigen Mechanismen entsprechen, die in derselben Richtung spielen., Wie auch immer, die Stabilitätsreihenfolge für Carbokationen sollte der Hyperkonjugation zugeschrieben werden (daher a+R-Verhalten auf einem freien p-Orbital, dem einfachsten π-System), obwohl auch andere Wechselwirkungen (wie Alkylpolarisierbarkeit) beteiligt sind (Aue, 2011).
Freie Radikale zeigen die gleiche Stabilitätsreihenfolge wie Carbokationen und weisen somit auf eine Stabilisierung durch Alkylsubstitution hin. Obwohl eine solche Stabilitätsreihenfolge auf der Grundlage eines angenommenen +I-Verhaltens gerechtfertigt sein kann, kann alternativ der +R-Effekt analog zur Stabilisierung freier Radikale durch einsame paartragende Atome betrachtet werden (Zipse, 2006).,
Reaktivität in Lösung
Relative Aziditäten einfacher Carbonsäuren in wässriger Lösung (Essigsäure>Propionsäure>Buttersäure) wurden in einigen Lehrbüchern verwendet, um den angenommenen +I-Effekt der Alkylgruppe zu veranschaulichen (Sorrell, 2006). Interessanterweise wird die umgekehrte Reihenfolge gefunden, wenn stattdessen Enthalpien betrachtet werden (Christensen, Izatt, & Hansen, 1967), was darauf hinweist, dass die Säurereihenfolge in wässriger Lösung Hydratationsentropien zugeschrieben werden sollte., Somit steht die signifikante Gitterreihenfolge von flüssigem Wasser (Verdampfungsentropie gleich 118.89 Jmol–1K–1, im Gegensatz zu typischen Werten von ca. 88Jmol-1K-1 für die meisten Flüssigkeiten, Dean, 1999) können erhebliche Änderungen an der Reaktionsenergie bewirken. Insbesondere die Hydratation apolarer Moleküle (oder Moi) führt zu einer weiteren Lösungsmittelgitterreihenfolge (Blokzijl & Engberts, 1993). Infolgedessen werden induktive Wirkungen von Alkylgruppen aus experimentellen Daten in wässriger Lösung häufig durch Hydratationsentropien maskiert (Calder & Barton, 1971)., Relative Aziditäten einfacher Carbonsäuren in der Gasphase (Yamdagni & Kebarle, 1973) und Acetonitril (Eckert et al., 2009) stimmen mit der Hauptrolle überein, die Hydratationsentropien spielen.
Der geringere Säuregehalt von Pivalsäure im Vergleich zu Essigsäure, der üblicherweise dem angenommenen +I-Effekt der Alkylgruppe zugeschrieben wird (Smith, 2008), wird umgekehrt, wenn Reaktionsenthalpien berücksichtigt werden (Eckert et al., 2009).,
Die angenommene + I Alkylgruppenwirkung auf den Säuregehalt einfacher Carbonsäuren in wässriger Lösung kann somit auf ein Artefakt zurückgeführt werden, das von Lösungsmitteleffekten abgeleitet ist. Während eine Volumenzunahme von neutralen gelösten Stoffen zu einer Hydratationsentropieerhöhung führt, findet sich die umgekehrte Beziehung für ionische Arten (Graziano, 2009). Infolgedessen führt die Alkylsubstitution (durch eine Erhöhung des Molekularvolumens) zur Stabilisierung (in Gibbs-freien Energiebedingungen) von nichtionisierter Säure in Wasser sowie zur Destabilisierung des entsprechenden Carboxylatanions, wodurch eine Säurerücknahme entsteht.,
Der größere Säuregehalt von Ameisensäure im Vergleich zu Essigsäure in wässriger Lösung (pKa-Werte: 3.751 bzw. 4.756, Dean, 1999) wurde auch in vielen Lehrbüchern als Beispiel für die Anwendung induktiver Effekte diskutiert (Hart, Hadad, Craine, & Hart, 2012; Hornback, 2006; Okuyama & Maskill, 2014; Roos & Roos, 2014). Da sehr ähnliche Reaktionsenthalpien an den Dissoziationsreaktionen von Ameisensäure und Essigsäure beteiligt sind (Christensen et al.,, 1967) muss der größere Säuregehalt der Ameisensäure tatsächlich auf Hydratationsentropieunterschiede zurückgeführt werden.
Schlussfolgerungen
Ein klares Verständnis der induktive-und Resonanz-Effekte ist ein wichtiger Schlüssel für ein gesundes lernen der Organischen Chemie (Mullins, 2008). Überraschenderweise wurde die fast allgegenwärtige Alkylgruppe in vielen Lehrbüchern fälschlicherweise als σ-Donor-Gruppe (+I) dargestellt. Ein duales Verhalten wird jedoch durch Alkylsubstituenten in Abhängigkeit von der Hybridisierung des Nachbaratoms gezeigt., Somit verhalten sich Alkylgruppen, die an aliphatische Ketten gebunden sind, als σ-Akzeptoren (–I, konsistent mit der größeren Elektronegativität von Kohlenstoff im Vergleich zu Wasserstoff), während diejenigen, die an π-Systeme gebunden sind, als π-Spender wirken (+R, aufgrund hyperkonjugativer Wechselwirkungen). Eine Reihe experimenteller und theoretischer Daten (Dipolmomente, NMR -, IR-und UV-Spektren, Reaktivität) stimmen mit einem solchen dualen Verhalten überein.,
Die gesamte Analyse aller hier betrachteten Daten erlaubt die Ableitung eines Small-I-Effekts sowie eines signifikanten + R-Verhaltens für die Alkylgruppe als Merkmal, das in allen Diskussionen über spektroskopische und Reaktivitätseigenschaften organischer Verbindungen gültig ist.
Interessenkonflikt
Der Autor erklärt keinen Interessenkonflikt.