introduktion
Substituenta effekter utgör ett nyckelbegrepp för förståelsen av reaktivitet och spektroskopiskt beteende hos organiska föreningar (Krygowski & St??pień, 2005). I ett enkelt tillvägagångssätt kan substituenteffekter klassificeras enligt interaktionsmekanismen med det reaktiva centret som induktivt (Genom σ-bindningar) eller resonanseffekter (Genom π-bindningar)., Vissa ytterligare termer (t.ex. steriska, fält-eller lösningsmedelseffekter) skulle dock krävas för en grundlig beskrivning av substituenteffekterna.
sedan Ingolds klassificering av elektroniska substituenteffekter (Ingold, 1953) har alkylgruppen betraktats som en σ-donatorsubstituent (+i, I Ingolds nomenklatur) i de flesta organiska Kemi läroböcker (Burrows, Holman, Parsons, Pilling, & pris, 2013; Hornback, 2006; Roos & Roos, 2014; Smith, 2013; vollhardt & Schore, 2014)., Eğes kritik mot en sådan förenklad synvinkel bör dock påpekas:
”i vatten är propansyra något svagare än ättiksyra. Naturen hos den induktiva effekten av en alkylgrupp debatteras av kemister. Alkylgrupper stabiliserar karbokationer och i den rollen verkar det vara elektronfrisättande. De ökar också aminernas grundlighet, vilket tyder på att de är elektronfrisättande. Å andra sidan, även om tert-butylalkohol (PKA 19) är en svagare syra än etanol (PKA 17) i vatten, är det starkare syra i gasfasen., Denna experimentella observation tyder på att alkylgrupper kan stabilisera anjoner såväl som katjoner och att solvering spelar en viktig roll för att bestämma relativa surheter. Således är ett varningens ord nödvändigt. De relativa aciditeterna som de generaliseringar som presenteras i detta kapitel baseras på bestämdes i vatten. I gasfasen ses ofta återföringar i storleksordningen besläktade föreningar.”(Eğe, 1999, s. 107)
vissa alkylersättningseffekter har ofta förklarats i läroböcker på motsägelsefulla eller gåtfulla sätt., Således tillskrivs kemiska skiftskillnader mellan CH3-och CH2-grupper i Hornbacks bok att ”kolet är något mer elektronegativt än väte” (Hornback, 2006, s. 549) trots att alkylgruppen tidigare har klassificerats som en svag induktiv elektrondonerande substituent (Hornback, 2006, s. 117). I vollhardts lärobok illustreras förhållandet mellan metylgruppens kemiska skift för ett antal CH3X-föreningar och X-elektronegativiteten med en tabell som saknar en post för X = metyl (Vollhardt & Schore, 2014, s., 389), vilket undviker den obekväma kolfrågan.
Jag visar här att alkylgruppen beter sig som A –i+R-substituent. Även om vissa faktorer (t.ex. fält -, sterik-eller lösningsmedelseffekter) implicit ignoreras i detta tillvägagångssätt, kan många tillgängliga teoretiska och experimentella bevis beskrivas på ett enkelt sätt.
en Cδ–Hδ+ bondpolarisering har experimentellt observerats för metan (lazzeretti, Zanasi, & Raynes, 1987), konsekvent med den större elektronegativiteten av kol i förhållande till väte, 2,55 vs 2.,20 i Pauling skala (Allred, 1961). Ett sådant polarisationsmönster gör det möjligt att förutsäga dipolmomentriktningen för enkla kolväten genom tillsatsmodeller, även om kvantitativt avtal vanligtvis är blygsamt (2-metylpropan: 0.3 d beräknad vs. 0.132 d experimentell) (Dean, 1999).
eftersom väte används som standard i Ingolds klassificering av substituenter (Krygowski & St??pień, 2005), alkylgruppen bör klassificeras som A-i-substituent (därmed en σ-elektronutdragande grupp). En sådan roll illustreras i Fig., 1 För σ-bindningspolariseringen från vilken atom Y som helst till en metylgrupp, även om omvänd bindningspolarisering förväntas när Y är en mer elektronegativ än kol (t.ex. klor).
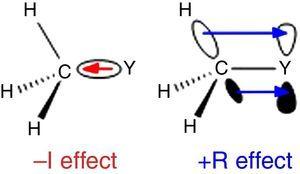
–i (vänster) och +R (höger) effekter av en metylgrupp bunden till en atom Y.
ett annat beteende finns för alkylgrupper när de är kopplade till sp2 eller sp-hybridiserade atomer på grund av elektrondensitetsdonation från alkyl C–H eller c–c σ-bindningar till den tomma p-orbitalen hos den angränsande atomen (det enklaste π-systemet), som visas i Fig. 1. Således kan minskningen av gasfasens surhet för fenol och bensoesyra genom p-metylsubstitution (McMahon & Kebarle, 1977) endast hänföras till en signifikant π-givareffekt för metylsubstituent (faktiskt större än för metoxigruppen)., Alkylgruppen bör dock betraktas som en atypisk π-donatorsubstituent på grund av bristen på ensamma elektronpar. En sådan σ-bond / π – systeminteraktion, namngiven som hyperkonjugation (Mullins, 2012) kan lätt förklaras analogt med π-donatorbeteendet hos en ensam parbärande atom (t.ex. klor) till en tom p–orbital, även om C–C eller C-H-bindningar (snarare än elektron lone par) i alkylgruppen är involverade som elektronfrisättande enheter i hyperkonjugativa interaktioner., Intressant är π→σ * interaktioner (negativ hyperkonjugation) vanligtvis försumbar för alkylgrupper som saknar elektronegativa atomer (Bocca, Pontes, & Basso, 2004).
vissa molekylära strukturella egenskaper kan rationaliseras på grundval av alkylgruppens egenskaper. Till exempel, de större Co bond längder som finns i metylketoner(aceton: exp. 1.210 Å, calc. 1.193 Å) i jämförelse med relaterade aldehyder (acetaldehyd: exp. 1.209 Å, calc. 1.,188Å) (Berry, Waltman, Pacansky, & Hagler, 1995) kan hänföras till stabiliseringen av zwitterionisk resonansform (se Fig. 2) genom alkylgrupp π-donation till karbonylkolatomen, vilket försvagar dubbelbindningsfunktionen hos karbonylgruppen.
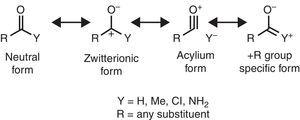
neutrala (vänster) och zwitterioniska (höger) resonansformer av en karbonylförening.
Hyperkonjugativa interaktioner är beroende av arrangemanget av C–H (eller C–C) bindningar i förhållande till p-orbitalen hos den angränsande atomen Y, den mest effektiva interaktionen som motsvarar ett nästan parallellt arrangemang. Till exempel är toluen csp3-H-bindningen nästan vinkelrät mot ramplanet något längre än de andra csp3–H-bindningarna (av 0.002 Å, Hameka & Jensen, 1996)., Geometrinberoende av hyperkonjugering gör det möjligt att förklara konformationsanalysen av metylsubstituerade omättade föreningar, såsom Propen (Liberles, O ’ Leary, Eilers, & Whitman, 1972) eller acetaldehyd (Muñoz-Caro, Niño, & Moule, 1994).
som en välkänd följd av alkylgruppens π-donatorbeteende ger alkylerubstitution mer elektronrika alkener och Aren (Libit & Hoffmann, 1974)., Den höga reaktiviteten hos en alkylsubstituerad Aren i en SEAr-reaktion kan sålunda hänföras till stabiliseringen av motsvarande Wheland-mellanliggande Genom π-elektrondonation.
alkylgruppens –i+R-beteende gör det möjligt att förklara ett antal egenskaper hos alkylsubstituerade föreningar, såsom dipolmoment, spektroskopiska egenskaper och reaktivitet (i gasfas och lösningsmedia), som visas nedan.
dipolmoment
alkylgruppens elektronutdragningsbeteende i alifatiska föreningar återspeglas också i dipolmoment., Således dipol ögonblick vektorer för propan och 2-metylpropan (Tasi et al., 1997), liksom vissa substituerade bicyklooktaner (Böhm & Exner, 2004) kan hänföras till metylgruppens återkallande effekt (–i) i jämförelse med väte (se Fig. 3).
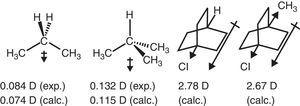
dipol stunder av propan och substituerade bicyklooktaner.
däremot krävs π-donatorns karaktär av metylgrupp (+R) för att förklara ökningen av dipolmoment av nitrobensen och bensonitril genom p-metylsubstitution (brun, 1959) (se Fig. 4).
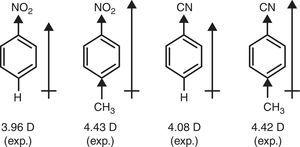
dipolmoment för bensenderivat.
molekylära dipolmoment kan beräknas på ett tillförlitligt sätt med nuvarande beräkningsmetoder., Intressant är de beräknade dipolmomentvektorerna för en uppsättning enkla kolväten (Tasi et al., 1997) har gjort det möjligt att dra slutsatsen en dubbel roll för metylgruppen: elektronutdragning när den är kopplad till sp3 – kolatomer, men elektrondonering när den är bunden till sp2 eller sp3-kol.
ett sådant dubbelbeteende hos alkylsubstituenten observeras också för heteroatombärande föreningar. Således observeras en gradvis dipolmomentminskning för successiv metylsubstitution på ammoniak (NH3, 1.47 D; MeNH2, 1.31 D; Me2NH, 1.01 d; Me3N, 0.,61 d) (Le Fèvre & Russell, 1947), i samförstånd med den progressiva minskningen av kväveelektrondensiteten (Hehre & Pople, 1970). Däremot finns en dipolmomentförbättring (från 1.53 d till 1.68 d) (Nelson, Lide, & Maryott, 1967) för N,N-dimetylsubstitution på anilin (Targema, Obi-Egbedi, & Adeoye, 2013), konsekvent med höjningen av π-donatorns karaktär för aminogruppen (Hinchliffe & Kidd, 1980) på grund av +r-bidrag från Metylsubstituenter.,
spektroskopiska egenskaper
spektroskopiska egenskaper hos många organiska föreningar kan lätt rationaliseras genom att anta A –I+R-beteende för alkylgruppen som en allmän egenskap. Således kan NMR-kemiska förskjutningen av en atom betraktas som ett experimentellt mått på elektrondensiteten vid motsvarande kärnposition, även om andra effekter – såsom anisotropa magnetfält – också kan vara involverade. Downfield Skift induceras av en metylsubstituent på sp3 kolatomer (+9,6 ppm i 13C NMR) eller motsvarande bundna väteatomer (+0.,63ppm i 1H NMR) (Pretsch, Bühlmann, & Badertscher, 2009) överensstämmer med beteendet hos typiska –i-grupper (såsom halogenatomer).
Alkylersättningseffekter på NMR kemiska skiftningar av alkener visar en minskning av elektrondensiteten i α-positionen (+12,9 ppm för 13C NMR; + 0,45 ppm för 1H NMR), liksom en täthetshöjning i β –positionen (-7,4 ppm för 13C; -0,31/-0,40 ppm för 1H), konsekvent med A-i+R-effekten, även om anisotropa effekter (såsom ringströmmar) också kan spela en roll. Sådant a-i+R-beteende finns också för alkyner, enligt 13C NMR-spektroskopi (+8.,5 ppm för α position, -3.6 ppm för β position).
det dikotomösa beteendet hos alkylsubstituenter på π-system (elektrondensitetshöjning för α-atom, elektrondensitetsminskning för β-atom) kan inte förklaras på grundval av ett enkelt beteende (t.ex. en +i-effekt).
–jag+R beteende (Meier, 2007) iakttas genom 15N NMR-spektroskopi för alkyl substitution på aminer och amider beroende på kväve hybridisering (downfield skift för alifatiska aminer, upfield skift för Nsp2 räntebärande föreningar – såsom anilines och amider).,
NMR-kopplingskonstanter är också beroende av substituenta elektroniska egenskaper (liksom vissa geometriska egenskaper). Således finns en signifikant minskning för 1H-1H kopplingskonstanter genom metylsubstitution (trans, -2,3 Hz; cis, -1,6 Hz; gem, -0,4 Hz), i kvalitativ överenskommelse med data från typiska elektronutdragande grupper, såsom fluoratom (trans, -6,3 Hz; cis, -6,9 Hz; gem, -5,7 Hz). Det positiva bidraget för metylsubstitution på 13C-1H kopplingskonstanter av alifatiska föreningar (+1.,0Hz), är också kvalitativt överensstämmer med de från andra –i grupper (fluor, +24Hz).
infraröd spektroskopi är också känslig för substituentegenskaper, vilket illustreras av co-sträckningsfrekvensen för karbonylföreningar som en funktion av motsvarande substituent Y, som kan rationaliseras i form av resonansformer (Fig. 2). Genom att ta en alifatisk aldehyd (ca., 1725cm–1) som referens kan redshift (wavenumber minskning) inducerad av A +i–substituent (acetyltrimetylsilan, 1645cm-1: Soderquist & Hsu, 1982) hänföras till stabiliseringen av zwitterionformen. Istället provocerade blueshift av A–i-substituent (acylklorider, >1800cm-1: Pretsch et al., 2009) kan förklaras med hjälp av två alternativa eller samtidiga mekanismer (destabilisering av zwitterionformen och/eller bidrag från en acyliumjonbärande form). Slutligen redshifts provocerade av + R substituenter (amides, ca., 1680cm–1: Pretsch et al., 2009) kan hänföras till bidraget från en specifik resonansform. Den lätta rödförskjutning som alkylgruppen inducerar (metylketoner, ca. 1715cm-1) visar en nettoelektrondonerande effekt (följaktligen en övervägande av +R –effekten över-i-egenskaperna). Nettodonatoreffekten av den karbonylbundna alkylgruppen överensstämmer med acetons större dipolmoment (2.88 D) i förhållande till formaldehyd (2.33 D) (Nelson et al., 1967).
alkylgruppens påverkan på UV–Vis-spektra av många föreningar kan också förklaras när det gäller elektroniska effekter., Således är badokroma Skift inducerade av alkylgrupper på UV-absorptionsband av α, β-omättade föreningar (+10nm i α-position, +12nm i β-position), konjugerade polyener (+5nm) eller bensenderivat (+3.0 nm) kvalitativt förenliga med effekterna av typiska π-donorgrupper (t.ex. klor).
gasfas syra–bas reaktivitet
relativa basiciteter av alifatiska aminer i vattenlösning har hänförts till den antagna +i-effekten av alkylgruppen (Sorrell, 2006)., Interestingly, the irregular basicity order of amines in water (Me2NH>MeNH2>Me3N>NH3, as shown by the pKa values for the corresponding conjugated acids: 10.77>10.62>9.80>9.,246) (Dean, 1999) är förorenat av lösningsmedelseffekter som illustreras av den systematiska basicitetsordningen av aminer i gasfasen (Me3N>Me2NH>menh2>NH3) (Brauman, Riveros, & Blair, 1971)., Även om gasfasens basicitetsordning kan hänföras till den vanligtvis antagna +i –alkyleffekten (Carter, 2007), har en minskning av kväveelektrondensiteten genom metylsubstitution faktiskt observerats med hjälp av molekylära elektrostatiska potentiella beräkningar (Baeten, de Proft, & Geerlings, 1995), vilket indikerar a-i-beteende för metylgruppen., Faktum är att gasfasbasicitetsordningen för alifatiska aminer bör hänföras till den ökande stabiliseringen av substituerade ammoniumjoner på grund av alkylgruppens polarisabilitet (Aue, Webb, & Bowers, 1976).
de relativa surheterna av alkoholer i vattenlösning (H2O>MeOH>EtOH>iPrOH>tBuOH) har också i vissa läroböcker tillskrivits den antagna alkyl +i-effekten (Johnson, 1999; solomoner, fryhle, & Snyder, 2016)., Eftersom den omvända surhetsordningen finns i gasfasen bör de relativa surheterna av alkoholer i vatten hänföras till de lägre magnituderna av solvationsintalpier för större alkoxidanjoner (Brauman & Blair, 1969).
diskussionen om alkylgruppens elektroniska egenskaper kan också tillämpas på karbanioner. Således har”läroboken”stabilitetsordern för enkla karbanioner (metyl>etyl>isopropyl>tert-butyl) hänförts till den antagna +i-induktiva effekten av alkylgrupper (Burrows et al., 2013; för Chaloner, 2015; Roos & Roos, 2014; Smith, 2013). En oregelbunden ordning finns dock för gasfas carbanion stabilities (tBu>Me>iPr>Et), i samförstånd med samspelet mellan två motsatta alkyleffekter (DePuy et al., 1989): en stabiliserande mekanism genom alkylpolarisabilitet (det vill säga n→σ* hyperkonjugation) och en destabiliserande trend (konsekvent med en +R-roll, genom att anta ett p-liknande beteende för carbon lone pair).,
stabiliteten hos andra reaktionsmellanprodukter kan också bedömas på grundval av alkylgruppseffekter. Således har den välkända stabilitetsordern för carbocations (tertiary>sekundär>primär>metyl) ibland tillskrivits en positiv induktiv effekt (Chaloner, 2015; Roos & Roos, 2014)., Intressant är hyperkonjugering presenteras i många läroböcker som en alternativ förklaring till stabiliteten för karbokationer (brun, Iverson, Anslyn, & Foote, 2013; Burrows et al., 2013) även om det vanliga tvetydiga skrivandet förhindrar att man kontrollerar om båda förklaringarna motsvarar antingen två olika beskrivningar av samma fenomen eller två samtidiga mekanismer som spelar i samma riktning., Hur som helst bör stabilitetsordern för karbokationer hänföras till hyperkonjugering (därmed ett+R-beteende på en ledig p-orbital, det enklaste π-systemet), även om andra interaktioner (såsom alkylpolarisabilitet) också är inblandade (Aue, 2011).
fria radikaler visar samma stabilitetsordning som karbokationer, vilket indikerar stabilisering genom alkylerubstitution. Även om en sådan stabilitetsordning kan motiveras på grundval av ett antaget +i-beteende, kan +R-effekten alternativt betraktas, analogt med stabiliseringen av fria radikaler av lone parbärande atomer (Zipse, 2006).,
reaktivitet i lösning
relativa surheter av enkla karboxylsyror i vattenlösning (ättiksyra>propionsyra>smörsyra) har använts i vissa läroböcker för att illustrera alkylgruppens antagna +i-effekt (Sorrell, 2006). Intressant är att omvänd ordning hittas när enthalpies istället betraktas (Christensen, Izatt, & Hansen, 1967), vilket indikerar att surhetsordningen i vattenlösning bör hänföras till hydratisering entropier., Således, den betydande gitter ordning av flytande vatten (förångning entropi lika 118.89 Jmol-1K-1, i motsats till typiska värden för ca. 88Jmol–1k–1 för de flesta vätskor, Dean, 1999) kan införa betydande förändringar på reaktionsenergin. I synnerhet leder hydrering av apolära molekyler (eller moieties) till en ytterligare lösningsmedelsgitterbeställning (Blokzijl & Engberts, 1993). Som en konsekvens maskeras alkylgruppsinduktiva effekter från experimentella data i vattenlösning ofta av hydratiseringsentropier (Calder & Barton, 1971)., Relativ acidities av enkla karboxylsyror i gasfas (Yamdagni & Kebarle, 1973) och acetonitril (Eckert et al., 2009) överensstämmer med den stora roll som hydration entropies spelar.
den lägre surheten hos pivalinsyra i jämförelse med ättiksyra, vanligtvis hänförlig till den antagna +i-effekten av alkylgruppen (Smith, 2008), vänds när reaktionsentalpier beaktas (Eckert et al., 2009).,
den antagna +i-alkylgruppseffekten på surheten hos enkla karboxylsyror i vattenlösning kan således hänföras till en artefakt som härrör från lösningsmedelseffekter. Medan en volymökning av neutrala lösningar leder till en hydratisering entropihöjning, finns det omvända förhållandet för Joniska arter (Graziano, 2009). Som en följd av detta leder alkylersättning (genom en ökning av molekylvolymen) till stabiliseringen (i Gibbs fria energitermer) av icke-joniserad syra i vatten samt destabiliseringen av motsvarande karboxylatanjon, vilket resulterar i en surhetsminskning.,
den större surheten av myrsyra i jämförelse med ättiksyra i vattenlösning (PKA-värden: 3.751 respektive 4.756, Dean, 1999) har också diskuterats i många läroböcker som ett exempel på tillämpningen av induktiva effekter (Hart, Hadad, Craine, & Hart, 2012; Hornback, 2006; Okuyama & Maskill, 2014; Roos & Maskill, 2014; Roos & Roos, 2014). Eftersom mycket liknande reaktion entalpies är involverade i dissociation reaktioner av myrsyra och ättiksyra (Christensen et al.,, 1967), den större surheten av myrsyra måste verkligen tillskrivas hydratisering entropi skillnader.
slutsatser
en tydlig förståelse av induktiva och resonanseffekter är en viktig nyckel för ett bra lärande av organisk kemi (Mullins, 2008). Förvånansvärt har den nästan allestädes närvarande alkylgruppen felaktigt presenterats i många läroböcker som en σ-donator (+i) – grupp. Ett dubbelt beteende visas emellertid av alkylsubstituenter beroende på hybridiseringen av grannatomen., Således uppträder alkylgrupper som är bundna till alifatiska kedjor som σ-acceptorer (–i, konsekvent med den större elektronegativiteten av kol i förhållande till väte), medan de som är kopplade till π-system fungerar som π-givare (+R, på grund av hyperkonjugativa interaktioner). Ett antal experimentella och teoretiska data (dipolmoment, NMR, IR och UV-spektra, reaktivitet) är överens med ett sådant dubbelt beteende.,
hela analysen av alla data som övervägs här möjliggör en liten –i-effekt samt ett signifikant +R-beteende för alkylgruppen som en funktion som gäller i alla diskussioner om spektroskopiska och reaktivitetsegenskaper hos organiska föreningar.
intressekonflikt
författaren förklarar ingen intressekonflikt.