wprowadzenie
efekty substytucyjne stanowią kluczową koncepcję dla zrozumienia reaktywności i zachowania spektroskopowego związków organicznych (Krygowski & St??pień, 2005). W prostym podejściu efekty substytucyjne można klasyfikować zgodnie z mechanizmem interakcji z centrum reaktywnym jako indukcyjne (poprzez wiązania σ) lub rezonansowe (poprzez wiązania π)., Niemniej jednak, niektóre dalsze terminy (takie jak sterylne, pole lub efekty rozpuszczalnika) byłyby wymagane do dokładnego opisu efektów substytucyjnych.
od czasu klasyfikacji efektów substytucji elektronicznych Ingolda (Ingold, 1953) Grupa alkilowa została uznana za substytut σ-dawcy (+I, w nomenklaturze Ingolda) w większości podręczników Chemii Organicznej (Burrows, Holman, Parsons, Pilling, & Price, 2013; Hornback, 2006; Roos & , Niemniej jednak należy zwrócić uwagę na krytykę Eğe wobec tak uproszczonego punktu widzenia:
„w wodzie kwas propanowy jest nieco słabszy niż kwas octowy. Charakter indukcyjnego efektu grupy alkilowej jest dyskutowany przez chemików. Grupy alkilowe stabilizują węglowodory i w tej roli wydają się uwalniać elektrony. Zwiększają również zasadowość Amin, ponownie sugerując, że uwalniają one elektrony. Z drugiej strony, chociaż alkohol tert-butylowy (pKa 19) jest słabszym kwasem niż etanol (PKA 17) w wodzie, jest silniejszym kwasem w fazie gazowej., Ta obserwacja eksperymentalna sugeruje, że grupy alkilowe mogą stabilizować aniony, a także kationy i że solwatacja odgrywa ważną rolę w określaniu względnych kwasowości. Dlatego konieczne jest słowo ostrożności. Względne kwasowości, na których opierają się uogólnienia przedstawione w tym rozdziale, zostały określone w wodzie. W fazie gazowej często obserwuje się odwrócenia w kolejności związków pokrewnych.”(Eğe, 1999, str. 107)
niektóre efekty substytucji alkilowej były często wyjaśniane w podręcznikach w sprzeczny lub enigmatyczny sposób., Tak więc, chemiczne różnice przesunięć między grupami CH3 i CH2 są przypisywane w książce Hornback ' a faktowi, że „węgiel jest nieco bardziej elektronegatywny niż wodór” (Hornback, 2006, s. 549), pomimo że grupa alkilowa została wcześniej sklasyfikowana jako słaby indukcyjny substytut oddający elektrony (Hornback, 2006, s. 117). W podręczniku Vollhardta zależność między przesunięciami chemicznymi grupy metylowej dla wielu związków CH3X i elektronegatywności X jest zilustrowana tabelą bez wpisu dla X = metyl (Vollhardt & Schore, 2014, s., 389), unikając w ten sposób niewygodnego problemu węgla.
pokazuję tutaj, że grupa alkilowa zachowuje się jak podstawnik a –I+R. Chociaż niektóre czynniki (takie jak efekty polowe, steryczne lub rozpuszczalnikowe) są domyślnie ignorowane w tym podejściu, wiele obecnie dostępnych dowodów teoretycznych i eksperymentalnych można więc opisać w łatwy sposób.
a cδ–Hδ+ polaryzacja wiązania została eksperymentalnie zaobserwowana dla metanu (Lazzeretti, Zanasi,& Raynes, 1987), konsekwentnie z większą elektroujemnością węgla w stosunku do wodoru, 2,55 vs.2.,20 w skali Paulinga (Allred, 1961). Taki wzór polaryzacji pozwala przewidzieć kierunek momentu dipolowego prostych węglowodorów za pomocą modeli addytywnych, choć porozumienie ilościowe jest zwykle skromne (2-metylopropan: 0,3 d szacowany vs.0,132 d eksperymentalny) (Dean, 1999).
ponieważ wodór jest stosowany jako standard w klasyfikacji podstawników Ingolda (Krygowski& St??pień, 2005), grupę alkilową należy klasyfikować jako podstawnik a-I (stąd σ Grupa wycofująca elektrony). Taka rola zilustrowana jest na Rys., 1 dla σ polaryzacji wiązania od dowolnego atomu Y do grupy metylowej, choć odwrotna polaryzacja wiązania jest oczekiwana, gdy Y jest bardziej elektronegatywny niż węgiel (np. chlor).
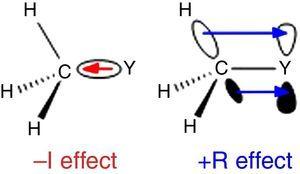
–i (po lewej) i +R (po prawej) wpływ grupy metylowej związanej z atomem Y.
inne zachowanie występuje dla grup alkilowych, gdy dołączone do SP2 lub sp-hybrydyzowanych atomów ze względu na donację gęstości elektronów z alkilowych wiązań C–H lub c–c σ do pustego orbitalu P przylegającego atomu (najprostszy układ π), Jak pokazano na Rys. 1. Tak więc spadek kwasowości w fazie gazowej fenolu i kwasu benzoesowego przez podstawienie P-metylowe (McMahon & Kebarle, 1977) można przypisać jedynie znaczącemu efektowi π-dawcy dla podstawnika metylowego (w rzeczywistości większy niż dla grupy metoksy)., Grupę alkilową należy jednak uznać za nietypowy substytut π-dawcy ze względu na brak par samotnych elektronów. Takie oddziaływanie σ-wiązania / π-układu, nazwane hiperkonjugacją (Mullins, 2012) może być łatwo wyjaśnione przez analogię z zachowaniem π-donora pojedynczego atomu (np. chloru) do pustego orbitala p, chociaż wiązania C-C lub C–H (zamiast samotnych par elektronowych) grupy alkilowej są zaangażowane jako jednostki uwalniające elektrony w hiperkonjugacyjnych oddziaływaniach., Co ciekawe, interakcje π → σ * (ujemna hiperkonjugacja) są zwykle nieistotne dla grup alkilowych pozbawionych atomów elektronegatywnych (Bocca, Pontes,& Basso, 2004).
niektóre molekularne cechy strukturalne można zracjonalizować na podstawie właściwości grupy alkilowej. Na przykład większe długości wiązania CO występujące w ketonach metylowych( aceton: exp. 1.210 Å, calc. 1.193 Å) w porównaniu z pokrewnymi aldehydami (aldehyd octowy: exp. 1.209 Å, calc. 1.,188å) (Berry, Waltman, Pacansky, & Hagler, 1995) można przypisać stabilizacji formy rezonansu zwitterionowego(patrz Rys. 2) przez grupę alkilową π-1 do atomu węgla karbonylowego, osłabiając w ten sposób funkcję wiązania podwójnego grupy karbonylowej.
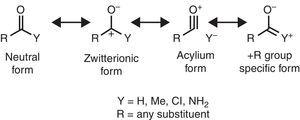
neutralne (z lewej) i zwitterionowe (z prawej) formy rezonansowe związku karbonylowego.
Hiperkonjugatywne interakcje zależą od ułożenia wiązań C–H (lub C–C) w stosunku do orbitalu P sąsiadującego atomu Y, najbardziej efektywnej interakcji odpowiadającej prawie równoległemu układowi. Na przykład Wiązanie toluenu Csp3–H prawie prostopadłe do płaszczyzny szkieletu jest nieco dłuższe niż inne wiązania Csp3-H (o 0,002 Å, Hameka & Jensen, 1996)., Zależność geometrii hiperkonjugacji pozwala wyjaśnić analizę konformacyjną związków nienasyconych podstawionych metylem, takich jak Propen (Liberles, O 'Leary, Eilers, & Whitman, 1972) lub aldehyd octowy (Muñoz-Caro, Niño, & Moule, 1994).
jako dobrze znana konsekwencja zachowania π-dawcy grupy alkilowej, podstawienie alkilowe daje więcej alkenów i arenów bogatych w elektrony (Libit & Hoffmann, 1974)., Wysoka reaktywność arenu podstawionego alkilem w reakcji SEAr może być zatem przypisana stabilizacji odpowiedniego półproduktu Whelanda poprzez oddawanie π-elektronów.
zachowanie –I+R grupy alkilowej pozwala wyjaśnić szereg cech związków podstawionych alkilami, takich jak momenty dipolowe, właściwości spektroskopowe i reaktywność (w fazie gazowej i roztworze), jak pokazano poniżej.
momenty dipolowe
zachowanie grupy alkilowej w związkach alifatycznych jest również odzwierciedlone w momentach dipolowych., Tak więc wektory momentu dipolowego dla propanu i 2-metylopropanu (Tasi et al., 1997), a także niektóre podstawione bicyklooktany (Böhm & Exner, 2004) można przypisać efektowi wycofania (- I) grupy metylowej w porównaniu z wodorem (patrz Rys. 3).
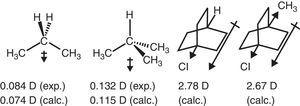
momenty dipolowe propanu i podstawionych bicyklooktanów.
w przeciwieństwie do tego, znak π-dawcy grupy metylowej (+R) jest wymagany w celu wyjaśnienia wzrostu momentów dipolowych nitrobenzenu i benzonitrylu poprzez podstawienie P-metylowe (Brown, 1959) (patrz Rys. 4).
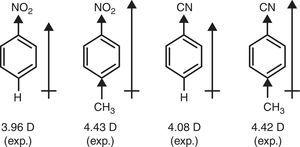
momenty dipolowe pochodnych benzenu.
momenty dipolowe molekularne mogą być niezawodnie obliczane za pomocą obecnych metod obliczeniowych., Co ciekawe, obliczone wektory momentu dipolowego dla zbioru prostych węglowodorów (Tasi et al., 1997) pozwoliły wywnioskować podwójną rolę grupy metylowej: wycofywanie elektronów przy przyłączaniu do atomów węgla sp3, ale oddawanie elektronów przy wiązaniu z węglami sp2 lub sp3.
takie podwójne zachowanie podstawnika alkilowego obserwuje się również dla związków heteroatomowych. W ten sposób obserwuje się stopniowe zmniejszanie momentu dipolowego w przypadku kolejnych podstawień metylowych amoniakiem (NH3, 1.47 D; MeNH2, 1.31 D; Me2NH, 1.01 d; Me3N, 0.,61 D) (Le Fèvre & Russell, 1947), w porozumieniu z postępującym zmniejszaniem gęstości elektronów azotu (Hehre & Pople, 1970). W przeciwieństwie do tego, wzmocnienie momentu dipolowego (od 1,53 D do 1,68 D) (Nelson, Lide, & Maryott, 1967) znajduje się dla substytucji n,n-dimetylu na anilinie (Targema, Obi-Egbedi, & Adeoye, 2013), konsekwentnie z podwyższeniem π-dawcy znak dla grupy aminowej (Hinchliffe &) ze względu na +r udział podstawników metylowych.,
właściwości spektroskopowe
właściwości spektroskopowe wielu związków organicznych można łatwo zracjonalizować, przyjmując zachowanie a-I+R dla grupy alkilowej jako ogólną cechę. Tak więc NMR przesunięcie chemiczne atomu może być traktowane jako eksperymentalna miara gęstości elektronowej w odpowiedniej pozycji jądra, choć inne efekty, takie jak anizotropowe pola magnetyczne, mogą być również zaangażowane. Przesunięcia w dół pola indukowane przez podstawnik metylowy na atomach węgla sp3 (+9,6 ppm w 13C NMR) lub odpowiadających im atomów wodoru (+0.,63ppm w 1H NMR) (Pretsch, Bühlmann, & Badertscher, 2009) są zgodne z zachowaniem typowych grup-I (takich jak atomy halogenowe).
wpływ podstawienia alkilowego na zmiany chemiczne NMR alkenów wykazuje spadek gęstości elektronów w pozycji α (+12,9 ppm dla 13C NMR; +0,45 ppm dla 1H NMR), a także wzrost gęstości w pozycji β (-7,4 ppm dla 13C; -0,31/-0,40 ppm dla 1H), konsekwentnie z efektem a –i+R, choć efekty anizotropowe (takie jak prądy pierścieniowe) mogą również odgrywać rolę. Takie zachowanie a-I+R występuje również dla alkinów, zgodnie ze spektroskopią 13C NMR (+8.,5ppm dla pozycji α, -3,6 ppm dla pozycji β).
dychotomiczne zachowanie podstawników alkilowych na układach π (wzrost gęstości elektronowej dla atomu α, spadek gęstości elektronowej dla atomu β) nie może być wyjaśnione na podstawie prostego zachowania (takiego jak efekt a +I).
zachowanie a –i+R (Meier, 2007) jest obserwowane przez spektroskopię 15N NMR dla podstawienia alkilowego na aminach i amidach w zależności od hybrydyzacji azotu (przesunięcia w dół pola dla Amin alifatycznych, przesunięcia w górę pola dla związków łożyskujących Nsp2-takich jak aniliny i amidy).,
stałe sprzężenia NMR są również zależne od podstawnikowych właściwości elektronicznych (a także niektórych cech geometrycznych). Tak więc, znaczny spadek znajduje Dla 1H–1H stałych sprzęgających poprzez podstawienie metylu (trans, -2,3 Hz; cis, -1,6 Hz; gem, -0,4 Hz), w jakościowej zgodzie z danymi z typowych grup wycofujących elektrony, takich jak atom fluoru (trans, -6,3 Hz; cis, -6,9 Hz; gem, -5,7 Hz). Dodatni udział w podstawieniu metylu na stałe sprzęgające związków alifatycznych 13C-1H (+1.,0Hz), jest również jakościowo zgodny z tymi z innych grup – i (fluor, +24Hz).
spektroskopia w podczerwieni jest również wrażliwa na właściwości podstawników, co ilustruje częstotliwość rozciągania związków karbonylowych jako funkcja odpowiedniego podstawnika Y, co można zracjonalizować w kategoriach form rezonansowych (rys. 2). Poprzez przyjmowanie aldehydu alifatycznego (ok., 1725cm-1) jako odniesienie, przesunięcie ku czerwieni (spadek liczby fal) wywołane przez podstawnik a +I (acetylotrimetylosilan, 1645cm–1: Soderquist & Hsu, 1982) można przypisać stabilizacji postaci zwitterionowej. Zamiast tego blueshift wywołany przez podstawnik a-I (chlorki acylu, >1800cm-1: Pretsch et al., 2009) można wyjaśnić za pomocą dwóch alternatywnych lub współbieżnych mechanizmów(destabilizacja postaci zwitterionowej i / lub udział postaci jonów acylium). W końcu przesunięcia Czerwieni wywołane przez podstawniki + R (amidy, ok., 1680cm-1: Pretsch et al., 2009) można przypisać wkładowi określonej formy rezonansowej. Lekkie przesunięcie ku czerwieni wywołane przez grupę alkilową (Ketony metylowe, ok. 1715cm-1) wykazuje efekt oddania elektronów netto (stąd przewaga efektu + R nad właściwościami-I). Efekt netto dawcy karbonylowej grupy alkilowej jest zgodny z większym momentem dipolowym acetonu (2,88 D) w stosunku do Formaldehydu (2,33 D) (Nelson et al., 1967).
wpływ grupy alkilowej na widmo UV–Vis wielu związków można również wyjaśnić w kategoriach efektów elektronicznych., Tak więc bathochromiczne przesunięcia indukowane przez grupy alkilowe na pasma absorpcji UV α,β-nienasyconych związków (+10 Nm w pozycji α, +12 Nm w pozycji β), sprzężonych polieenów (+5 Nm) lub pochodnych benzenu (+3,0 nm) są jakościowo zgodne z efektami typowych grup π-donorowych (np. chloru).
reaktywność fazy gazowej kwasowo–zasadowa
względne zasadowość Amin alifatycznych w roztworze wodnym przypisano zakładanemu efektowi + I grupy alkilowej (Sorrell, 2006)., Interestingly, the irregular basicity order of amines in water (Me2NH>MeNH2>Me3N>NH3, as shown by the pKa values for the corresponding conjugated acids: 10.77>10.62>9.80>9.,246) (Dean, 1999) jest zanieczyszczony przez działanie rozpuszczalników, jak ilustruje systematyczna kolejność zasadowości amin w fazie gazowej (Me3N>Me2NH>MeNH2>NH3) (Brauman, Riveros, & , Chociaż kolejność zasadowości fazy gazowej można przypisać Zwykle zakładanemu efektowi alkilowemu + I (Carter, 2007), zmniejszenie gęstości elektronów azotu przez podstawienie metylowe zostało rzeczywiście zaobserwowane za pomocą molekularnych obliczeń potencjału elektrostatycznego (Baeten, de Proft,& Geerlings, 1995), co wskazuje na zachowanie a –I dla grupy metylowej., W rzeczywistości kolejność zasadowości fazy gazowej Amin alifatycznych powinna być przypisana rosnącej stabilizacji podstawionych jonów amonowych ze względu na polaryzowalność grupy alkilowej (Aue, Webb, & Bowers, 1976).
względne kwasowości alkoholi w roztworze wodnym (H2O>MeOH>EtOH>iPrOH>tBuOH) mają w niektórych podręcznikach przypisywano również zakładany efekt alkil +i (Johnson, 1999; Solomons, fryhle,& Snyder, 2016)., Ponieważ odwrotna kolejność kwasowości znajduje się w fazie gazowej, względne kwasowości alkoholi w wodzie należy przypisać niższym magnitudom entalpii solwatacyjnych dla większych anionów alkooksydowych (Brauman & Blair, 1969).
dyskusja na temat własności elektronowych grupy alkilowej może dotyczyć również karbanionów. Tak więc „podręcznikowy” porządek stabilności dla prostych karbanionów (metylu>etylu>izopropylu>tert-butylu) został przypisany zakładanemu efektowi indukcyjnemu +I grup alkilowych (Burrows et al.,, 2013; Chaloner, 2015; Roos & Roos, 2014; Smith, 2013). Jednakże, nieregularny porządek znajduje się dla gazowej stabilności karbanionu (tBu>Me >iPr>Et), w porozumieniu z zbieżnością dwóch przeciwstawnych efektów alkilowych (DePuy et al., 1989): mechanizm stabilizujący poprzez polaryzację alkilową (tj. hiperkonjugację n → σ*) i trend destabilizujący (konsekwentnie z rolą a + R, przyjmując zachowanie podobne do p dla pary węglowej).,
stabilność innych półproduktów reakcji można również ocenić na podstawie efektów grupy alkilowej. Tak więc dobrze znany porządek stabilności dla karbokacji (trzeciorzędowy>wtórny>pierwotny>metylowy) jest czasami przypisywany dodatniemu efektowi indukcyjnemu (Chaloner, 2015; Roos & Roos, 2014)., Co ciekawe, hiperkonjugacja jest przedstawiona w wielu podręcznikach jako alternatywne Wyjaśnienie kolejności stabilności karbokacji (Brown, Iverson, Anslyn, & Foote, 2013; Burrows et al., 2013), choć zwykłe dwuznaczne pismo uniemożliwia ustalenie, czy oba wyjaśnienia odpowiadają albo dwóm różnym opisom tego samego zjawiska, albo dwóm współbieżnym mechanizmom grającym w tym samym kierunku., W każdym razie, kolejność stabilności dla karbokacji powinna być przypisana hiperkonjugacji (stąd zachowanie a + R na wolnym orbitalu P, najprostszym układzie π), chociaż inne interakcje (takie jak polaryzowalność alkilowa) są również zaangażowane (Aue, 2011).
wolne rodniki wykazują ten sam porządek stabilności co karbokacje, co wskazuje na stabilizację przez podstawienie alkilowe. Chociaż taki porządek stabilności może być uzasadniony na podstawie założonego zachowania +I, efekt + R może być alternatywnie traktowany, analogicznie do stabilizacji wolnych rodników przez pojedyncze atomy nośne (zipse, 2006).,
reaktywność w roztworze
względna kwasowość prostych kwasów karboksylowych w roztworze wodnym (kwas octowy>kwas propionowy> kwas masłowy) zostały wykorzystane w niektórych podręcznikach do zilustrowania zakładanego efektu +I grupy alkilowej (Sorrell, 2006). Co ciekawe, odwrotna kolejność znajduje się, gdy entalpy są traktowane zamiast (Christensen, Izatt, & Hansen, 1967), wskazując tym samym, że porządek kwasowości w roztworze wodnym powinien być przypisany entropii hydracji., Tak więc znaczący porządek sieciowy ciekłej wody (Entropia parowania równa 118,89 Jmol-1K-1, w przeciwieństwie do typowych wartości ok. 88Jmol-1K-1 dla większości cieczy, Dean, 1999) może wprowadzić znaczne zmiany w energetyce reakcji. W szczególności, uwodnienie cząsteczek apolarnych (lub cząsteczek) prowadzi do dalszego uporządkowania siatki rozpuszczalnika (Blokzijl & Engberts, 1993). W konsekwencji, indukcyjne efekty grupy alkilowej z danych doświadczalnych w roztworze wodnym są często maskowane przez entropie uwodnienia (Calder & Barton, 1971)., Względna kwasowość prostych kwasów karboksylowych w fazie gazowej (Yamdagni & Kebarle, 1973) i acetonitrylu (Eckert et al., 2009) są zgodne z główną rolą entropii hydratacyjnej.
niższa kwasowość kwasu piwalowego w porównaniu z kwasem octowym, zwykle przypisywana zakładanemu efektowi + I grupy alkilowej (Smith, 2008), jest odwrócona, gdy rozważa się entalpie reakcji (Eckert et al., 2009).,
zakładany wpływ grupy alkilowej +I na kwasowość prostych kwasów karboksylowych w roztworze wodnym można zatem przypisać artefaktowi uzyskanemu z efektów rozpuszczalnikowych. Podczas gdy wzrost objętości neutralnych roztworów prowadzi do wzrostu entropii hydratacyjnej, odwrotna zależność występuje dla gatunków jonowych (Graziano, 2009). W konsekwencji podstawienie alkilowe (poprzez zwiększenie objętości cząsteczkowej) prowadzi do stabilizacji (w kategoriach energii wolnej Gibbsa) niezjonizowanego kwasu w wodzie, a także destabilizacji odpowiadającego mu anionu karboksylowego, co powoduje spadek kwasowości.,
większa kwasowość kwasu mrówkowego w porównaniu z kwasem octowym w roztworze wodnym (wartości pKa: 3.751 i 4.756, odpowiednio, Dean, 1999) była również omawiana w wielu podręcznikach jako przykład zastosowania efektów indukcyjnych (Hart, Hadad, Craine, & Hart, 2012; Hornback, 2006; Okuyama & maskill, 2014; Roos & Roos, 2014). Ponieważ bardzo podobne reakcje entalpie biorą udział w reakcjach dysocjacji kwasów mrówkowego i octowego (Christensen et al.,, 1967), większą kwasowość kwasu mrówkowego należy rzeczywiście przypisać różnicom entropii hydratacyjnej.
wnioski
jasne zrozumienie efektów indukcyjnych i rezonansowych jest głównym kluczem do zdrowego uczenia się Chemii Organicznej (Mullins, 2008). Co zaskakujące, prawie wszechobecna Grupa alkilowa została błędnie przedstawiona w wielu podręcznikach jako grupa σ-dawcy (+I). Jednak podwójne zachowanie jest wykazywane przez podstawniki alkilowe w zależności od hybrydyzacji sąsiedniego atomu., Tak więc grupy alkilowe związane z łańcuchami alifatycznymi zachowują się jak σ-akceptory (- I, zgodnie z większą elektroujemnością węgla w stosunku do wodoru), podczas gdy te przyłączone do układów π działają jak π-donory (+R, ze względu na hiperkonjugatywne interakcje). Wiele danych eksperymentalnych i teoretycznych (momenty dipolowe, widma NMR, IR i UV, reaktywność) zgadza się z takim podwójnym zachowaniem.,
Cała analiza wszystkich analizowanych tutaj danych pozwala wnioskować o małym efekcie –I, jak również znaczącym zachowaniu +R dla grupy alkilowej jako Cechie ważnej we wszystkich dyskusjach na temat właściwości spektroskopowych i reaktywności związków organicznych.
konflikt interesów
Autor deklaruje brak konfliktu interesów.