Inleiding
Substituent effecten vormen een sleutelbegrip voor het begrijpen van reactiviteit en spectroscopisch gedrag van organische verbindingen (Krygowski & St??pień, 2005). In een eenvoudige benadering kunnen substituente effecten worden geclassificeerd volgens het mechanisme van interactie met het reactieve centrum als inductieve (Door σ-bindingen) of resonantie-effecten (door π-bindingen)., Niettemin zouden enkele verdere termen (zoals sterische, veld-of oplosmiddeleffecten) vereist zijn voor een grondige beschrijving van substituente effecten.sinds de Ingold ’s classification of electronic substituent effects (Ingold, 1953) wordt de alkylgroep beschouwd als een σ-donor substituent (+I, in de inGold’ s nomenclature) in de meeste organische chemie studieboeken (Burrows, Holman, Parsons, Pilling, &Price, 2013; Hornback, 2006; Roos& Roos, 2014; Smith, 2013; vollhardt& Schore, 2014)., Niettemin moet de kritiek van de Eğe op een dergelijk simplistisch standpunt worden opgemerkt:
“in water is propaanzuur iets zwakker dan azijnzuur. De aard van het inductieve effect van een alkylgroep wordt besproken door chemici. Alkylgroepen stabiliseren carbokaties en lijken in die rol elektron-vrijgevend te zijn. Ze verhogen ook de basiciteit van amines, wat weer suggereert dat ze elektronen vrijgeven. Aan de andere kant, hoewel tert-butylalcohol (PKA 19) een zwakker zuur is dan ethanol (PKA 17) in water, is het sterker zuur in de gasfase., Deze experimentele observatie suggereert dat alkylgroepen zowel anionen als kationen kunnen stabiliseren en dat solvatie een belangrijke rol speelt bij het bepalen van relatieve zuurheden. Dus een woord van voorzichtigheid is noodzakelijk. De relatieve zuurgraad waarop de in dit hoofdstuk gepresenteerde veralgemeningen zijn gebaseerd, werd bepaald in water. In de gasfase, worden omkeringen in de Orde van verwante samenstellingen vaak gezien.”(Eğe, 1999, p. 107)
sommige alkylsubstitutie-effecten zijn vaak in leerboeken op tegenstrijdige of raadselachtige manieren uitgelegd., Zo worden chemische verschuivingsverschillen tussen CH3 en CH2 groepen in het boek van de Hornback toegeschreven aan het feit dat “koolstof iets meer elektronegatief is dan waterstof” (Hornback, 2006, p. 549) ondanks dat de alkylgroep eerder is geclassificeerd als een zwak inductief elektron-donerende substituent (Hornback, 2006, p. 117). In het handboek van Vollhardt wordt de relatie tussen chemische verschuivingen in de methylgroep voor een aantal ch3x-verbindingen en de x-elektronegativiteit geïllustreerd door een tabel zonder vermelding voor X = methyl (Vollhardt & Schore, 2014, p., 389), waardoor het lastige koolstofprobleem wordt vermeden.
i laat hier zien dat de alkylgroep zich gedraagt als A-I+R substituent. Hoewel sommige factoren (zoals veld -, sterische of oplosmiddeleffecten) impliciet worden genegeerd in deze benadering, kunnen veel van de momenteel beschikbare theoretische en experimentele bewijzen dus op een eenvoudige manier worden beschreven.
a cδ — Hδ+ bond polarisatie is experimenteel waargenomen voor methaan (Lazzeretti, Zanasi, & Raynes, 1987), consistent met de grotere elektronegativiteit van koolstof ten opzichte van waterstof, 2,55 vs.2.,20 in the Pauling scale (Allred, 1961). Een dergelijk polarisatiepatroon maakt het mogelijk om de dipoolmomentrichting van eenvoudige koolwaterstoffen te voorspellen door middel van additieve modellen, hoewel kwantitatieve overeenkomst meestal bescheiden is (2-methylpropaan: 0,3 d geschat vs.0,132 d experimenteel) (Dean, 1999).
aangezien waterstof als standaard wordt gebruikt in de Ingold ’s classification of substituents (Krygowski & St??pień, 2005) moet de alkylgroep worden geclassificeerd als A –I-substituent (vandaar een σ-elektron-terugtrekkende groep). Een dergelijke rol wordt geïllustreerd in Fig., 1 Voor De σ binding polarisatie van welk atoom dan ook Y aan een methylgroep, hoewel de omgekeerde binding polarisatie wordt verwacht wanneer Y meer elektronegatief is dan koolstof (bijvoorbeeld chloor).
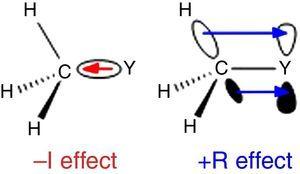
– I (links) en + R (rechts) effecten van een methylgroep gebonden aan een atoom Y.
een ander gedrag wordt gevonden voor alkylgroepen wanneer deze aan SP2 of SP-gehybridiseerde atomen zijn bevestigd door elektronendensiteitsdonatie van alkyl–C–H-of c-c-σ-bindingen met de lege p-orbitaal van het aan elkaar grenzende atoom (het eenvoudigste π-systeem), zoals weergegeven in Fig. 1. De afname van de zuurgraad in de gasfase voor fenol en benzoëzuur door P-methylsubstitutie (McMahon & Kebarle, 1977) kan dus alleen worden toegeschreven aan een significant π-donoreffect voor methylsubstituent (inderdaad groter dan dat Voor methoxygroep)., De alkylgroep moet echter worden beschouwd als een atypische π-donorsubstituent vanwege het ontbreken van eenzame elektronenparen. Een dergelijke σ-binding / π-systeem interactie, genoemd als hyperconjugatie (Mullins, 2012) kan gemakkelijk worden verklaard door analogie met het π-donorgedrag van een eenzaam paardragend atoom (bijvoorbeeld chloor) naar een lege p orbitaal, hoewel C-C of C–H bindingen (in plaats van elektron eenzame paren) van de alkylgroep betrokken zijn als elektron–bevrijdende eenheden in hyperconjugatieve interacties., Interessant is dat π→σ * interacties (negatieve hyperconjugatie) meestal verwaarloosbaar zijn voor alkylgroepen zonder elektronegatieve atomen (Bocca, Pontes, & Basso, 2004).
sommige moleculaire structurele kenmerken kunnen worden gerationaliseerd op basis van de eigenschappen van de alkylgroep. Bij voorbeeld de grotere Co-Bindingslengten die in methylketonen (aceton: exp. 1.210 Å, calc. 1,193 Å) in vergelijking met de verwante aldehyden (aceetaldehyde: exp. 1.209 Å, calc. 1.,188Å) (Berry, Waltman, Pacansky, & Hagler, 1995) kan worden toegeschreven aan de stabilisatie van de zwitterionische resonantievorm (zie Fig. 2) door alkylgroep π-donatie aan het carbonyl koolstofatoom, waardoor de dubbele binding van de carbonylgroep wordt verzwakt.
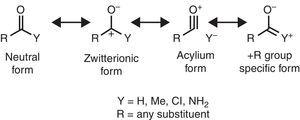
neutrale (links) en zwitterionische (rechts) resonantievormen van een carbonylverbinding.
Hyperconjugatieve interacties zijn afhankelijk van de rangschikking van C–H (of C–C) bindingen ten opzichte van de p-orbitaal van het aangrenzende atoom Y, de meest effectieve interactie die overeenkomt met een bijna parallelle rangschikking. De tolueen-csp3-H-binding, die bijna loodrecht staat op het kadervlak, is bijvoorbeeld iets langer dan de andere Csp3–H-bindingen (met 0,002 Å, Hameka & Jensen, 1996)., De geometrieafhankelijkheid van hyperconjugatie verklaart de conformationele analyse van methyl-gesubstitueerde onverzadigde verbindingen, zoals propeen (Liberles, O ‘ Leary, Eilers, & Whitman, 1972) of aceetaldehyde (Muñoz-Caro, Niño, & Moule, 1994).
als een bekend gevolg van het π-donorgedrag van de alkylgroep levert alkylsubstitutie meer elektronenrijke alkenen en arenes op (Libit & Hoffmann, 1974)., De hoge reactiviteit van een alkyl-gesubstitueerd arene in een Searreactie kan dus worden toegeschreven aan de stabilisatie van het overeenkomstige Wheland-tussenproduct Door π-elektrondonatie.
het –I + R-gedrag van de alkylgroep maakt het mogelijk een aantal kenmerken van alkyl-gesubstitueerde verbindingen te verklaren, zoals dipoolmomenten, spectroscopische eigenschappen en reactiviteit (in gasfase en oplossingsmedia), zoals hieronder weergegeven.
Dipoolmomenten
het elektron-terugtrekkende gedrag van alkylgroep in alifatische verbindingen wordt ook weerspiegeld in dipoolmomenten., Dus, de dipoolmoment vectoren voor propaan en 2-methylpropaan (Tasi et al., 1997), evenals enkele gesubstitueerde bicyclooctanen (Böhm & Exner, 2004) kunnen worden toegeschreven aan het terugtrekkende effect (–I) van de methylgroep in vergelijking met waterstof (zie Fig. 3).
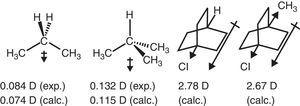
Dipoolmomenten van propaan en gesubstitueerde bicyclooctanen.
daarentegen is het π-donorkarakter van methylgroep (+R) vereist om de toename van dipoolmomenten van nitrobenzeen en benzonitril door P-methylsubstitutie te verklaren (Brown, 1959) (zie Fig. 4).
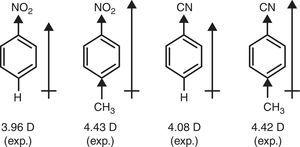
dipoolmomenten van benzeenderivaten.
moleculaire dipoolmomenten kunnen betrouwbaar worden berekend met behulp van de huidige rekenmethoden., Interessant is dat de berekende dipoolmomentvectoren voor een verzameling eenvoudige koolwaterstoffen (Tasi et al.(1997) hebben een dubbele rol voor de methylgroep laten afleiden: elektron-terugtrekkend wanneer gehecht aan sp3 koolstofatomen, maar elektron-donerend wanneer gebonden aan SP2 of sp3 koolstofatomen.
een dergelijk duaal gedrag van de alkylsubstituent wordt ook waargenomen voor heteroatoomhoudende verbindingen. Zo wordt een geleidelijke afname van het dipoolmoment waargenomen voor opeenvolgende methylsubstitutie op ammoniak (NH3, 1,47 D; MeNH2, 1,31 D; Me2NH, 1,01 D; Me3N, 0.,61 D) (Le Fèvre & Russell, 1947), in agreement with the progressive diminution of the nitrogen electron density (Hehre & Pople, 1970). In tegenstelling, een dipool moment van de verhoging (van 1.53 D te 1.68 D) (Nelson, Lide, & Maryott, 1967) is gevonden voor N,N-dimethyl substitutie op aniline (Targema, Obi-Egbedi, & Adeoye, 2013), in overeenstemming met het verhogen van de π-donor-teken voor de amino-groep (Hinchliffe & Kidd, 1980) door +R bijdragen van methyl substituenten.,
spectroscopische eigenschappen
spectroscopische eigenschappen van veel organische verbindingen kunnen gemakkelijk worden gerationaliseerd door het A –I+R-gedrag voor de alkylgroep als algemeen kenmerk aan te nemen. Aldus, kan de chemische verschuiving van NMR van een atoom als een experimentele maat van de elektronendichtheid bij de overeenkomstige kernpositie worden beschouwd hoewel andere gevolgen – zoals anisotrope magnetische velden – ook kunnen worden betrokken. Downfield verschuivingen veroorzaakt door een methylsubstituent op sp3 koolstofatomen (+9,6 ppm in 13C NMR) of de overeenkomstige gebonden waterstofatomen (+0.,63ppm in 1H NMR) (Pretsch, Bühlmann, & Badertscher, 2009) zijn consistent met het gedrag van typische –I-groepen (zoals halogeenatomen).
Alkyl substitutie-effecten op de NMR chemische verschuivingen van alkenen toon een elektron dichtheid daling in α-positie (+12.9 ppm voor 13C NMR; +0.45 ppm voor 1H NMR), evenals een dichtheid verhogen in β-positie (-7.4 ppm voor 13C; -0.31/-0.40 ppm voor 1H), consistent met een –I+R-effect, maar anisotrope effecten (zoals ring stromen) kunnen ook een rol spelen. Dergelijk a-I+R gedrag wordt ook gevonden voor alkynen, volgens 13C NMR spectroscopie (+8.,5ppm Voor α-positie, -3,6 ppm voor β-positie).
het dichotoom gedrag van alkylsubstituenten op π-systemen (verhoging van de elektronendichtheid Voor α-atoom, vermindering van de elektronendichtheid voor β-atoom) kan niet worden verklaard op basis van een eenvoudig gedrag (zoals A +I-effect).
A –I+R gedrag (Meier, 2007) wordt waargenomen door middel van 15N NMR spectroscopie voor alkylsubstitutie op amines en amiden afhankelijk van de stikstofhybridisatie (downfield shifts voor alifatische amines, upfield shifts voor nsp2-dragende verbindingen – zoals anilines en amiden).,
NMR-koppelingsconstanten zijn ook afhankelijk van substituente elektronische eigenschappen (evenals enkele geometrische kenmerken). Zo wordt een significante daling gevonden voor 1h-1H koppelingsconstanten door methylsubstitutie (trans, -2,3 Hz; cis, -1,6 Hz; gem, -0,4 Hz), in kwalitatieve overeenstemming met gegevens van typische elektron-onttrekkende groepen, zoals het fluor-atoom (trans, -6,3 Hz; cis, -6,9 Hz; gem, -5,7 Hz). De positieve bijdrage voor methylsubstitutie op 13C-1H koppelingsconstanten van alifatische verbindingen (+1.,0Hz), is ook kwalitatief consistent met die van andere –I groepen (fluor, +24Hz).
infraroodspectroscopie is ook gevoelig voor substituenteigenschappen, zoals wordt geïllustreerd door de co-rekfrequentie van carbonylverbindingen als functie van de overeenkomstige substituent Y, die kan worden gerationaliseerd in termen van resonantievormen (Fig. 2). Door het nemen van een alifatisch aldehyde (ca., 1725cm-1) als referentie kan de roodverschuiving (wavenumber afname) veroorzaakt door een +I substituent (acetyltrimethylsilaan, 1645cm–1: Soderquist & Hsu, 1982) worden toegeschreven aan de stabilisatie van de zwitterionische vorm. In plaats daarvan wordt de blauwverschuiving veroorzaakt door A-I substituent (acylchloriden, >1800cm-1: Pretsch et al., 2009) kan worden verklaard aan de hand van twee alternatieve of gelijktijdige mechanismen (destabilisatie van de zwitterionische vorm en/of bijdrage van een acylium-Ion-dragende vorm). Tot slot, de roodverschuivingen veroorzaakt door + R substituenten (amides, ca., 1680cm-1: Pretsch et al., 2009) kan worden toegeschreven aan de bijdrage van een specifieke resonantievorm. De lichte roodverschuiving veroorzaakt door alkylgroep (methylketonen, ca. 1715cm-1) toont een netto elektron-donerende effect (vandaar, een overwicht van het + R effect boven – i eigenschappen). Het netto donoreffect van de carbonylgebonden alkylgroep komt overeen met het grotere dipoolmoment van aceton (2,88 D) ten opzichte van formaldehyde (2,33 D) (Nelson et al., 1967).
de invloed van de alkylgroep op de UV-Vis-spectra van vele verbindingen kan ook worden verklaard in termen van elektronische effecten., Zo zijn de door alkylgroepen geïnduceerde veranderingen in de UV-absorptiebanden van α,β-onverzadigde verbindingen (+10 nm In α-positie, +12 nm In β-positie), geconjugeerde polyenen (+5 Nm) of benzeenderivaten (+3,0 nm) kwalitatief consistent met de effecten van typische π-donorgroepen (bijv. chloor).
gasfase zuur–base reactiviteit
relatieve basiciteiten van alifatische aminen in waterige oplossing zijn toegeschreven aan het veronderstelde +I effect van alkylgroep (Sorrell, 2006)., Interestingly, the irregular basicity order of amines in water (Me2NH>MeNH2>Me3N>NH3, as shown by the pKa values for the corresponding conjugated acids: 10.77>10.62>9.80>9.,246) (Dean, 1999) is verontreinigd door solventeffecten zoals geïllustreerd door de systematische basiciteitsvolgorde van amines in gasfase (Me3n>Me2NH>MeNH2>NH3) (Brauman, Riveros, &
* Blair, 1971)., Hoewel de basiciteitsvolgorde van de gasfase kan worden toegeschreven aan het gewoonlijk veronderstelde +I alkyleffect (Carter, 2007), is een afname van de stikstofelektrondichtheid door methylsubstitutie inderdaad waargenomen door middel van moleculaire elektrostatische Potentiaalberekeningen (Baeten, De Proft, & Geerlings, 1995), wat wijst op a –I gedrag voor de methylgroep., In feite moet de gasfasebasiciteitsorde van alifatische aminen worden toegeschreven aan de toenemende stabilisatie van gesubstitueerde ammoniumionen als gevolg van de polariseerbaarheid van de alkylgroep (Aue, Webb, & Bowers, 1976).
in vergelijking acidities van alcoholen in waterige oplossing (H2O>MeOH>EtOH>iPrOH>tBuOH) hebben toegeschreven in sommige leerboeken voor de verwachte alkyl +I-effect (Johnson, 1999; Solomons, Fryhle, & Snyder, 2016)., Aangezien de omgekeerde volgorde van de zuurgraad in de gasfase wordt gevonden, moeten de relatieve zuurgraad van alcoholen in water worden toegeschreven aan de lagere magnitudes van oplos-enthalpieën voor grotere alkoxide-anionen (Brauman & Blair, 1969).
de discussie over de elektronische eigenschappen van alkylgroepen kan ook worden toegepast op carbanionen. Zo is de stabiliteitsvolgorde voor enkelvoudige carbanionen (methyl>ethyl>isopropyl>tert-butyl) toegeschreven aan het veronderstelde +I inductief effect van alkylgroepen (Burrows et al.,, 2013; Chaloner, 2015; Roos & Roos, 2014; Smith, 2013). Er wordt echter een onregelmatige volgorde gevonden voor de stabiliteit van gasfasecarbanion (tBu>Me>iPr>Et), in overeenstemming met het samenvallen van twee tegengestelde alkyleffecten (DePuy et al., 1989): een stabiliserend mechanisme door alkylpolariseerbaarheid (dat wil zeggen, n→σ* hyperconjugatie) en een destabiliserende trend (consistent met A +R rol, door een P-achtig gedrag aan te nemen voor het koolstof eenzame paar).,
de stabiliteit van andere reactie-tussenproducten kan ook worden beoordeeld op basis van alkylgroepeffecten. De bekende stabiliteitsvolgorde voor carbocaties (tertiair>secundair>primair>methyl) wordt soms toegeschreven aan een positief inductief effect (Chaloner, 2015; Roos & Roos, 2014)., Interessant is dat hyperconjugatie in veel studieboeken wordt gepresenteerd als een alternatieve verklaring voor de stabiliteitsorde van carbocaties (Brown, Iverson, Anslyn, & Foote, 2013; Burrows et al., 2013) hoewel het gebruikelijke dubbelzinnige schrijven verhindert vast te stellen of beide verklaringen overeenkomen met twee verschillende beschrijvingen van hetzelfde fenomeen of twee gelijktijdige mechanismen die in dezelfde richting spelen., Hoe dan ook, de stabiliteitsorde voor carbokaties moet worden toegeschreven aan hyperconjugatie (vandaar A+R-gedrag op een leeg p-orbitaal, het eenvoudigste π-systeem), hoewel ook andere interacties (zoals alkylpolariseerbaarheid) betrokken zijn (Aue, 2011).
vrije radicalen vertonen dezelfde stabiliteitsvolgorde als carbokaties, wat duidt op stabilisatie door alkylsubstitutie. Hoewel een dergelijke stabiliteitsorde kan worden gerechtvaardigd op basis van een aangenomen +I-gedrag, kan het +R-effect als alternatief worden beschouwd, analoog aan de stabilisatie van vrije radicalen door eenzame paardragende atomen (Zipse, 2006).,
reactiviteit in oplossing
relatieve zuurgraad van enkelvoudige carbonzuren in waterige oplossing (azijnzuur>propionzuur>boterzuur) is gebruikt in sommige handboeken om het veronderstelde +I effect van de alkylgroep te illustreren (Sorrell, 2006). Interessant is dat de omgekeerde volgorde wordt gevonden wanneer enthalpieën in plaats daarvan worden beschouwd (Christensen, Izatt, & Hansen, 1967), wat erop wijst dat de volgorde van de zuurgraad in waterige oplossing moet worden toegeschreven aan hydratatie-entropieën., Zo is de significante roosterorde van vloeibaar water (verdamping entropie gelijk aan 118.89 Jmol–1K–1, in tegenstelling tot typische waarden van ca. 88Jmol-1K-1 voor de meeste vloeistoffen, Dean, 1999) kan aanzienlijke veranderingen in de reactie-energetica introduceren. In het bijzonder leidt hydratatie van apolaire moleculen (of delen) tot een verdere ordening van het solventrooster (Blokzijl & Engberts, 1993). Als gevolg hiervan worden inductieve effecten van alkylgroepen uit experimentele gegevens in waterige oplossing vaak gemaskeerd door hydratatie-entropieën (Calder & Barton, 1971)., Relatieve zuurgraad van enkelvoudige carbonzuren in gasfase (Yamdagni & Kebarle, 1973) en acetonitril (Eckert et al., 2009) consistent zijn met de belangrijke rol die hydratatie-entropieën spelen.
de lagere zuurgraad van pivalinezuur in vergelijking met azijnzuur, gewoonlijk toegeschreven aan het veronderstelde +I-effect van de alkylgroep (Smith, 2008), wordt omgekeerd wanneer rekening wordt gehouden met reactie-enthalpieën (Eckert et al., 2009).,
het veronderstelde + I alkylgroep-effect op de zuurgraad van enkelvoudige carbonzuren in waterige oplossing kan dus worden toegeschreven aan een artefact dat is afgeleid van oplosmiddeleffecten. Terwijl een volumetoename van neutrale opgeloste stoffen leidt tot een hydratatie entropie verhoging, wordt de omgekeerde relatie gevonden voor Ionische soorten (Graziano, 2009). Als gevolg hiervan leidt alkylsubstitutie (door een toename van het moleculaire volume) tot de stabilisatie (in termen van Gibbs vrije energie) van niet-geïoniseerd zuur in water en tot de destabilisatie van het overeenkomstige carboxylaatanion, waardoor de zuurgraad afneemt.,
de grotere zuurgraad van mierenzuur in vergelijking met azijnzuur in waterige oplossing (pKa-waarden: 3.751 en 4.756, respectievelijk, Dean, 1999) is ook besproken in veel studieboeken als een voorbeeld van de toepassing van inductieve effecten (Hart, Hadad, Craine, & Hart, 2012; Hornback, 2006; Okuyama & Maskill, 2014; roos & roos, 2014). Aangezien zeer gelijkaardige reactie enthalpieën betrokken zijn bij de dissociatiereacties van mierenzuur en azijnzuur (Christensen et al.,, 1967), de grotere zuurgraad van mierenzuur moet inderdaad worden toegeschreven aan hydratatie entropie verschillen.
conclusies
een duidelijk begrip van inductieve en resonantie-effecten is een belangrijke sleutel voor een goed leren van Organische Chemie (Mullins, 2008). Verrassend genoeg is de bijna alomtegenwoordige alkylgroep in veel handboeken ten onrechte gepresenteerd als een σ-donor (+I) groep. Nochtans, wordt een dubbel gedrag getoond door alkyl substituents afhankelijk van de kruising van het buuratoom., Zo gedragen alkylgroepen gebonden aan alifatische ketens zich als σ-acceptoren (–I, consistent met de grotere elektronegativiteit van koolstof ten opzichte van waterstof), terwijl die verbonden aan π-systemen fungeren als π-donors (+R, als gevolg van hyperconjugatieve interacties). Een aantal experimentele en theoretische gegevens (dipoolmomenten, NMR, IR, en UV spectra, reactiviteit) zijn het eens met een dergelijk duaal gedrag.,
de hele analyse van alle hier onderzochte gegevens maakt het mogelijk om een klein I –effect en een significant +R-gedrag voor de alkylgroep te concluderen als een kenmerk dat geldig is in alle discussies over spectroscopische en reactiviteitseigenschappen van organische verbindingen.
belangenverstrengeling
de auteur verklaart geen belangenverstrengeling.