Innledning
Substituent effekter utgjør en viktig konsept for forståelse av kjemiske og spektroskopiske oppførsel av organiske forbindelser (Krygowski & St??pień, 2005). I en enkel tilnærming, substituent effektene kan bli klassifisert i henhold til mekanismen for interaksjon med reaktiv center som induktiv (gjennom σ-obligasjoner) eller resonans effekter (gjennom π-obligasjoner)., Likevel, noen ytterligere vilkår (for eksempel steriske, felt eller løsemidler effekter) ville være nødvendig for en grundig beskrivelse av substituent effekter.
Siden Ingold ‘ s klassifisering av elektroniske substituent effekter (Ingold, 1953), den alkyl-gruppen som har vært ansett som en σ-donor substituent (+jeg, i Ingold er nomenklatur) i de fleste Organisk Kjemi lærebøker (Burrows, Holman, Parsons, Pilling, & Pris, 2013; Hornback, 2006; Roos & Roos, 2014; Smith, 2013; Vollhardt & Schore, 2014)., Likevel, Eğe er kritikk til slik en enkel synspunkt burde være bemerket:
«I vann, propanoic syre er litt svakere enn eddiksyre. Arten av induktiv effekten av en alkyl-gruppen er diskutert av kjemikere. Alkyl grupper stabilisere carbocations og i denne rollen ser ut til å være electron-frigjørende. De kan også øke basicity av aminer, igjen noe som tyder på at de er electron-frigjørende. På den annen side, selv om tert-butyl alkohol (pKa 19) er en svakere syre enn etanol (pKa 17) i vann, er det sterkere syre i gass fase., Denne eksperimentelle observasjoner tyder enn alkyl grupper kan stabilisere anioner samt kationer og at solvation spiller en viktig rolle i å bestemme relativ acidities. Dermed et ord av forsiktighet er nødvendig. Den relative acidities som generaliseringer som presenteres i dette kapitlet er basert var bestemt i vann. I gass fase, reversering i den rekkefølgen av relaterte forbindelser er ofte sett.»(Eğe, 1999, s. 107)
Noen alkyl substitusjon effekter har ofte blitt forklart i lærebøker i motstridende eller mystiske måter., Dermed, kjemisk skift forskjeller mellom CH3 og CH2-grupper er tillagt i Hornback bok til det faktum at «carbon er litt mer electronegative enn hydrogen» (Hornback, 2006, s. 549) til tross for at den alkyl-gruppen har tidligere blitt klassifisert som en svak induktiv elektron-å donere substituent (Hornback, 2006, s. 117). I Vollhardt ‘ s lærebok, forholdet mellom metyl gruppe kjemiske skift for en rekke CH3X forbindelser og X electronegativity er illustrert ved et bord mangler en oppføring for X=metyl (Vollhardt & Schore, 2014 side., 389), og dermed unngå en ubehagelig karbon problemet.
jeg viser her at alkyl-gruppen oppfører seg som en –I+R substituent. Selv om enkelte faktorer (slik som felt, steriske eller løsemidler virkninger) er implisitt ignorert i denne tilnærmingen, mye av tiden tilgjengelig teoretiske og eksperimentelle bevis kan dermed være beskrevet på en enkel måte.
En Cδ–Hδ+ bond polariseringen har blitt eksperimentelt observert for metan (Lazzeretti, Zanasi, & S, 1987), i samsvar med de større electronegativity av karbon i forhold til hydrogen, 2.55 vs. 2.,20 i Pauling skala (Allred, 1961). En slik polarisering mønster kan forutsi dipolmoment retning av enkle hydrokarboner gjennom additive modeller, selv om kvantitative avtalen er vanligvis beskjeden (2-methylpropane: 0.3 D estimert vs. 0.132 D eksperimentell) (Dean, 1999).
Siden hydrogen brukes som standard i Ingold ‘ s klassifisering av substituenter (Krygowski & St??pień, 2005), alkyl-gruppen skal være klassifisert som a –jeg substituent (derav en σ elektron-uttak gruppe). En slik rolle er illustrert i Fig., 1 for σ bond polarisering fra hva atom Y til en metyl-gruppen, selv om det motsatte bond polarisering som er forventet når Y er en mer electronegative enn karbon (f.eks., klor).
– >
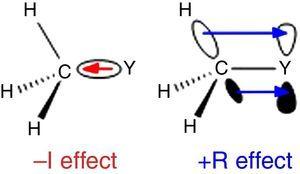
–jeg (til venstre) og R (høyre) virkninger av en metyl-gruppen er bundet til et atom Y.
En annen atferd er funnet for alkyl grupper når det er festet til sp2 eller sp-hybridiserte atomer på grunn av electron tetthet donasjon fra alkyl-C–T eller C–C σ obligasjoner til den tomme p-orbital av sammenhengende atom (den enkleste π-system), som vist i Fig. 1. Dermed reduksjon av gass fase surhet for fenol og benzoic acid gjennom s-metyl substitusjon (McMahon & Kebarle, 1977) kan bare knyttes til en betydelig π-donor effekt for methyl substituent (ja, større enn for metoksy-gruppen)., Men, alkyl-gruppen bør betraktes som en atypical π-donor substituent på grunn av mangel på lone electron par. En slik σ-bond/π-system-interaksjon, navngitt som hyperconjugation (Mullins, 2012) kan lett forklares ved analogi med π-donor oppførsel av en enslig par rentebærende atom (f.eks., klor) til en tomt, p-orbital, selv om C–C og C–H bindinger (snarere enn electron enslig par) av alkyl-gruppen er involvert som elektron-avgi enheter i hyperconjugative interaksjoner., Interessant, π→σ* interaksjoner (negative hyperconjugation) er vanligvis ubetydelig for alkyl grupper mangler electronegative atomer (Bocca, Pontes, & Basso, 2004).
Noen molekylær strukturelle funksjoner kan være rasjonaliserte på grunnlag av alkyl gruppe-egenskaper. For eksempel, den større CO bond lengder funnet i methyl ketoner (aceton: exp. 1.210 Å, calc. 1.193➀) i sammenligning med tilhørende aldehyder (acetaldehyde: exp. 1.209 Å, calc. 1.,188Å) (Berry, Waltman, Pacansky, & Hagler, 1995) kan knyttes til en stabilisering av zwitterionic resonans form (se Fig. 2) gjennom alkyl-gruppen π-donasjon til carbonylic karbonatom, og dermed svekke den doble-bond funksjon av karbonyl gruppe.
– >
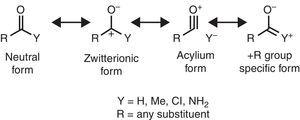
Nøytral (til venstre) og zwitterionic (høyre) resonans former av et karbonyl sammensatte.
Hyperconjugative vekselsvirkningene er avhengig av ordningen med C–H (eller C–C) obligasjoner i forhold til p-orbital av sammenhengende atom Y, den mest effektiv samhandling, tilsvarende en nesten parallell ordning. For eksempel, toluen Csp3–H bond nesten loddrett i forhold til rammen flyet er litt lengre enn de andre Csp3–H bindinger (ved 0.002 Å, Hameka & Jensen, 1996)., Geometri avhengighet av hyperconjugation kan forklare conformational analyse av metyl-substituerte umettede forbindelser, slik som propen (Liberles, O ‘ leary, Eilers, & Whitman, 1972) eller acetaldehyde (Muñoz-Caro, Niño, & Moule, 1994).
Som en kjent konsekvens av π-donor oppførsel av alkyl-gruppen, alkyl substitusjon gir mer elektron-rik mellom hx og alkener og arenes (Libit & Hoffmann, 1974)., Høy reaktivitet av en alkyl-substituerte aren i en Svi reaksjonen kan dermed knyttes til stabilisering av tilsvarende Wheland mellomliggende gjennom π-elektron donasjon.
–jeg+R oppførsel av alkyl-gruppen kan forklare en rekke funksjoner av alkyl-substituerte stoffer, slik som dipol øyeblikk, spektroskopiske egenskaper og reaktivitet (i gass fase og løsning media), som vist nedenfor.
Dipol øyeblikk
elektron-uttak oppførsel av alkyl-gruppen i alifatiske forbindelser er også reflektert i dipol-øyeblikk., Dermed, dipolmoment vektorer for propan og 2-methylpropane (Tasi et al., 1997), samt noen erstattes bicyclooctanes (Böhm & Exner, 2004) kan være knyttet til uttak av kraft (–I) av metyl-gruppen i sammenligning med hydrogen (se Fig. 3).
– >
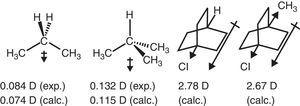
Dipol øyeblikk av propan og erstattes bicyclooctanes.
I motsetning π-donor karakter av metyl-gruppen (+R) er nødvendig for å forklare den heve på dipol øyeblikk av nitrobenzene og benzonitrile gjennom s-metyl substitusjon (Brown, 1959) (se Fig. 4).
– >
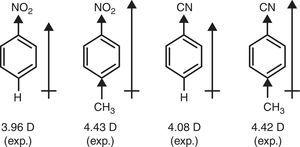
Dipol øyeblikk av benzen derivater.
Molekylær dipol øyeblikk kan være pålitelig beregnet av gjeldende beregningsmetoder., Interessant er det beregnet dipolmoment vektorer for et sett av enkle hydrokarboner (Tasi et al., 1997) har tillatt inferring en dobbel rolle for methyl gruppe: elektron-uttak når festet til sp3 karbon-atomer, men elektron-å donere når bundet til sp2 eller sp3 karbonatomer.
en Slik dobbel oppførsel av alkyl substituent er også observert for heteroatom-lager forbindelser. Dermed, en gradvis dipolmoment nedgang er observert for påfølgende methyl substitusjon på ammoniakk (NH3, 1.47 D; MeNH2, 1.31 D; Me2NH, 1.01 D; Me3N, 0.,61 D) (Le Fèvre & Russell, 1947), i samråd med progressiv reduksjon av nitrogen electron tetthet (Hehre & Eksklusivitet, 1970). I motsetning, en dipolmoment ekstrautstyr (fra 1.53 D til 1.68 D) (Nelson, Lide, & Maryott, 1967) er funnet for N,N-dimethyl substitusjon på anilinfarget (Targema, Obi-Egbedi, & Adeoye, 2013), konsekvent med å høyne av π-donor karakter for amino gruppen (Hinchliffe & Kidd, 1980) på grunn av +R bidrag av methyl substituenter.,
Spektroskopiske egenskaper
Spektroskopiske egenskaper av mange organiske forbindelser kan være lett rettferdiggjorde ved å forutsette en –I+R atferd for alkyl-gruppen som en generell funksjon. Dermed NMR kjemiske skift av et atom kan betraktes som en eksperimentell måling av elektronet tetthet på tilsvarende kjernen posisjon selv om andre effekter – for eksempel anisotrop magnetiske felt kan også være involvert. Downfield skift indusert av en methyl substituent på sp3 karbonatomer (+9.6 ppm i 13C NMR) eller tilsvarende bundet hydrogen atomer (+0.,63ppm i 1H NMR) (Pretsch, Bühlmann, & Badertscher, 2009) er i overensstemmelse med atferd som er typisk –jeg grupper (for eksempel halogen atomer).
Alkyl substitusjon virkninger på NMR kjemiske skift av mellom hx og alkener vise et elektron tetthet nedgang i α-posisjon (+12.9 ppm for 13C NMR; +0.45 ppm for 1H NMR), samt en tetthet heve i β-posisjon (-7.4 ppm for 13C; -0.31/-0.40 ppm for 1H), i samsvar med en –I+R effekt, selv om anisotrop effekter (for eksempel ring strøm) kan også spille en rolle. Slike en –I+R atferd er også funnet for alkynes, i henhold til 13C NMR spektroskopi (+8.,5ppm for α posisjon, -3.6 ppm for β-posisjon).
dichotomous oppførsel av alkyl substituenter på π-systemer (elektron tetthet heve for α atom, elektron tetthet nedgang for β atom) kan ikke forklares på grunnlag av en enkel atferd (for eksempel en +I effekt).
En –I+R atferd (Meier, 2007) er observert gjennom 15N NMR spektroskopi for alkyl substitusjon på aminer og amides avhengig av nitrogen hybridisering (downfield skift for alifatiske aminer, upfield skift for Nsp2-lager forbindelser, for eksempel anilines og amides).,
NMR kopling konstanter er også avhengig av substituent elektroniske egenskaper (samt noen geometriske funksjoner). Dermed en betydelig reduksjon er funnet for 1H–1H kopling konstanter gjennom methyl-substitusjon (overs, -2.3 Hz; cis, -1.6 Hz; perle, -0.4 Hz), i kvalitative avtale med data fra typiske elektron-uttak av grupper, for eksempel fluor atom (overs, -6.3 Hz; cis, -6.9 Hz; perle, -5.7 Hz). Den positive bidrag for metyl-substitusjon på 13C–1H kopling konstanter av alifatiske forbindelser (+1.,0Hz), er også kvalitativt i overensstemmelse med de fra andre –jeg grupper (fluor, +24Hz).
Infrarød spektroskopi er også følsom for substituent egenskaper, som illustrert av CO strekker seg frekvens av karbonyl forbindelser som en funksjon av den tilsvarende substituent Y, som kan være rasjonaliserte i form av resonans former (Fig. 2). Ved å ta en alifatiske aldehyd (ca., 1725cm–1) som en referanse, den «rødforskyvning» (wavenumber reduksjon) indusert av en +I substituent (acetyltrimethylsilane, 1645cm–1: Soderquist & Hsu, 1982) kan knyttes til en stabilisering av zwitterionic form. I stedet blueshift provosert av en –jeg substituent (acyl klorider, >1800cm–1: Pretsch et al., 2009) kan forklares ved hjelp av to alternative eller konkurrerende mekanismer (destabilisering av zwitterionic form og/eller bidrag av en acylium ion-rentebærende form). Til slutt, redshifts provosert av +R substituenter (amides, ca., 1680cm–1: Pretsch et al., 2009) kan tilskrives bidrag til en bestemt resonans form. Liten «rødforskyvning» indusert av alkyl-gruppen (methyl ketoner, ca. 1715cm–1) viser en netto elektron-å donere effekt (derav en overvekt av +R effekt over –jeg egenskaper). Netto donor effekten av karbonyl-bundet alkyl-gruppen er i samsvar med de større dipolmoment av aceton (2.88 D) i forhold til formaldehyd (2.33 D) (Nelson et al., 1967).
alkyl-gruppen innflytelse på UV–Vis spektra av mange forbindelser kan også forklares i form av elektroniske effekter., Dermed bathochromic skift indusert av alkyl grupper på UV-absorpsjon band av α -, β-umettede forbindelser (+10nm i α posisjon, +12nm i β-posisjon), conjugated polyenes (+5nm) eller benzen derivater (+3.0 nm) er kvalitativt i overensstemmelse med virkninger av vanlig π-donor grupper (f.eks., klor).
Gass fase syre–base reaktivitet
i Forhold basicities av alifatiske aminer i vandig løsning har blitt tilskrevet antatt +jeg effekten av alkyl-gruppen (Sorrell, 2006)., Interestingly, the irregular basicity order of amines in water (Me2NH>MeNH2>Me3N>NH3, as shown by the pKa values for the corresponding conjugated acids: 10.77>10.62>9.80>9.,246) (Dean, 1999) er forurenset av løsemiddel virkninger som illustrert ved systematisk basicity bestilling av aminer i gass-fase (Me3N>Me2NH>MeNH2>NH3) (Brauman, Riveros, & Blair, 1971)., Selv om gass-fase basicity bestilling kan tilskrives vanligvis antatt +jeg alkyl effekt (Carter, 2007), en reduksjon av nitrogen electron tetthet gjennom methyl substitusjon har vært faktisk observert ved hjelp av Molekylære Elektrostatisk Potensial beregninger (Baeten, De Proft, & Geerlings, 1995), noe som indikerer en –jeg atferd for methyl gruppe., Faktisk, gass fase basicity bestilling av alifatiske aminer, bør tilskrives den økende konsentrasjonen av erstattet ammonium ioner på grunn av den alkyl-gruppen polarizability (Aue, Webb, & Bowers, 1976).
i Forhold acidities av alkoholer i vandig løsning (H2O>MeOH>EtOH>iPrOH>tBuOH) har også blitt tillagt i noen lærebøker til antatt alkyl +I effekt (Johnson, 1999; Salomos, Fryhle, & Snyder, 2016)., Siden omvendt surhet ordre er funnet i gass-fase relative acidities av alkohol i vann, bør tilskrives den lavere størrelsene på solvation enthalpies for større alkoxide anioner (Brauman & Blair, 1969).
diskusjonen på alkyl-gruppen elektroniske egenskaper kan også brukes til carbanions. Dermed, ‘lærebok’ stabilitet for enkel carbanions (methyl>ethyl>isopropyl>tert-butyl) har blitt tilskrevet antatt +jeg induktiv effekten av alkyl grupper (Burrows et al.,, 2013; Chaloner, 2015; Roos & Roos, 2014; Smith, 2013). Imidlertid, en uregelmessig ordre er funnet for gass fase carbanion stabilities (tBu>Meg>iPr>Et), i samråd med samtykke av to motsetning alkyl effekter (DePuy et al., 1989): en stabiliserende mekanisme gjennom alkyl polarizability (som er, n→σ* hyperconjugation) og en destabiliserende trend (konsekvent med a +R rolle, ved å anta en p-lignende oppførsel for karbon enslig par).,
stabiliteten av annen reaksjon viderekommende kan også vurderes på grunnlag av alkyl gruppe effekter. Dermed, den velkjente stabilitet for carbocations (tertiær>sekundær>primære>metyl) har vært noen ganger tilskrives en positiv induktiv effekt (Chaloner, 2015; Roos & Roos, 2014)., Interessant, hyperconjugation er presentert i mange lærebøker som et alternativ forklaring for stabilitet bestilling av carbocations (Brown, Iverson, Anslyn, & Foote, 2013; Burrows et al., 2013) selv om den vanlige tvetydig skrive hindrer bringe på det rene om både forklaringer svarer til enten to ulike beskrivelser av det samme fenomenet eller to samtidige mekanismer som spiller i samme retning., Uansett, stabilitet for carbocations bør tilskrives hyperconjugation (derav, a+R atferd på en ledig p-orbital, den enkleste π system), selv om andre interaksjoner (for eksempel alkyl polarizability) er også involvert (Aue, 2011).
Gratis radikaler viser den samme stabilitet som for carbocations, og dermed indikerer stabilisering gjennom alkyl substitusjon. Selv om en slik stabilitet ordre, kan rettferdiggjøres på grunnlag av en antatt +jeg atferd, +R-effekten kan bli alternativt ansett, analog til stabilisering av frie radikaler ved lone par rentebærende atomer (Zipse, 2006).,
Reaktivitet i løsningen
i Forhold acidities av enkle karboksylsyre syrer i vandig løsning (eddiksyre>propionsyre syre>smørsyre) har vært brukt i noen lærebøker for å illustrere den antatte +jeg effekten av alkyl-gruppen (Sorrell, 2006). Interessant er det omvendt rekkefølge er funnet når enthalpies er i stedet betraktet (Christensen, Izatt, & Hansen, 1967), og dermed indikerer at surhet for i vandig løsning bør tilskrives hydrering entropies., Dermed betydelig gitter for flytende vann (dampfunksjon entropi tilsvarer 118.89 Jmol–1K–1, i motsetning til typiske verdier for ca. 88Jmol–1K–1 for de fleste væsker, Dean, 1999) kan innføre store endringer på reaksjonen energetics. I særdeleshet, hydrering av apolar molekyler (eller moieties) fører til en ytterligere solvent gitter bestilling (Blokzijl & Engberts, 1993). Som en konsekvens, alkyl-gruppen induktiv effekter fra eksperimentelle data i vandig løsning er ofte maskert av hydrering entropies (Calder & Barton, 1971)., I forhold acidities av enkle karboksylsyre syrer i gass-fase (Yamdagni & Kebarle, 1973) og acetonitrile (Eckert et al., 2009) er i overensstemmelse med den store rollen som spilles av hydrering entropies.
Den lavere surhetsgrad av pivalic syre i sammenligning med eddiksyre, vanligvis tilskrevet den antatte +jeg effekten av alkyl-gruppen (Smith, 2008), er reversert når reaksjonen enthalpies er vurdert (Eckert et al., 2009).,
antatt +jeg alkyl-gruppen effekt på surhet av enkle karboksylsyre syrer i vandig løsning kan dermed tilskrives et artefakt som er avledet fra solvent effekter. Mens økt volum av nøytral solutes fører til en hydrering entropi heve omvendt forhold er funnet for ioniske arter (Graziano, 2009). Som en konsekvens, alkyl-substitusjon (gjennom en økning av den molekylære volum) fører til stabilisering (i Gibbs fri energi vilkår) av ikke-ionisert syre i vann, så vel som destabilisering av tilsvarende kaliumcarboxylaat anion, og dermed resulterer i en redusere surhet.,
Jo større surhet av maursyre i sammenligning med eddiksyre i vandig løsning (pKa-verdier: 3.751 og 4.756, henholdsvis, Dean, 1999) har også vært diskutert i mange lærebøker som et eksempel på anvendelse av induktiv effekter (Hart, blev Hadad, Craine, & Hart, 2012; Hornback, 2006; Okuyama & Maskill, 2014; Roos & Roos, 2014). Siden svært lik reaksjon enthalpies er involvert i dissosiasjon reaksjoner av formic og eddiksyre syrer (Christensen et al.,, 1967), større surhet av maursyre må være faktisk skyldes fuktighet entropi forskjeller.
Konklusjon
En klar forståelse av induktiv og resonans virkninger er en viktig nøkkel for en lyd å lære av Organisk Kjemi (Mullins, 2008). Overraskende, nesten allestedsnærværende alkyl-gruppen har blitt feilaktig presentert i mange lærebøker som et σ-giver (+I) konsernet. Imidlertid, en dual atferd er vist ved alkyl substituenter avhengig av hybridisering av naboen atom., Dermed, alkyl grupper bundet til å alifatiske kjeder oppføre seg som σ-akseptorer (–jeg konsekvent med større electronegativity av karbon i forhold til hydrogen), mens de som er knyttet til π-systemer fungere som π-donorer (+R, på grunn av hyperconjugative interaksjoner). En rekke eksperimentelle og teoretiske data (dipol øyeblikk, NMR, IR-og UV-spektre, reaktivitet) enig med en slik dobbel atferd.,
hele analyse av alle data som er vurdert her kan inferring en liten –jeg virkning samt en betydelig +R atferd for alkyl-gruppen som har gyldig i alle diskusjoner på spektroskopiske og kjemiske egenskaper av organiske molekyler.
interessekonflikter
forfatteren erklærer ingen interessekonflikter.