はじめに
置換基の効果は、有機化合物の反応性と分光挙動の理解のための重要な概念を構成する(Krygowski&St??パイエ、2005年)。 単純なアプローチでは、置換基効果は、反応中心との相互作用のメカニズムに従って誘導(π結合を介して)または共鳴効果(π結合を介して)として分類することができる。, それにもかかわらず、置換基効果の完全な説明のためには、いくつかのさらなる用語(立体効果、場効果または溶媒効果など)が必要であろう。
Ingoldの電子置換基効果の分類以来(Ingold、1953)、アルキル基は、ほとんどの有機化学の教科書(Burrows、Holman、Parsons、Pilling、&Price、2013;Hornback、2006;Roos&Roos、2014;Smith、2013;vollhardt&schore,2014)., それにもかかわらず、このような単純な視点に対するEßeの批判は注目されるべきである:
“水中では、プロパン酸は酢酸よりもわずかに弱い。 アルキル基の誘導効果の性質は、化学者によって議論されている。 アルキル基はカルボカチオンを安定化し,その役割で電子放出しているように見える。 それらはまた、アミンの塩基度を増加させ、再びそれらが電子放出していることを示唆している。 一方、tert-ブチルアルコール(pKa19)は水中ではエタノール(pKa17)よりも弱い酸であるが、気相ではより強い酸である。, この実験的観察は,アルキル基よりも陽イオンと同様に陰イオンを安定化でき,溶媒和が相対酸度を決定する上で重要な役割を果たすことを示唆している。 したがって、注意の言葉が必要です。 この章で提示された一般化が基づいている相対的な酸性度は、水中で決定された。 気相では、関連する化合物の順に反転することがしばしば見られる。”(Eşe,1999,p.107)
いくつかのアルキル置換効果は、しばしば矛盾したまたは謎めいた方法で教科書で説明されている。, したがって、CH3基とCH2基の化学シフトの違いは、アルキル基が以前に弱い誘導電子供与性置換基として分類されていたにもかかわらず、”炭素は水素よりもわずかに電気陰性である”(Hornback、2006、p.549)という事実に起因する(Hornback、2006、p.117)。 Vollhardtの教科書では、いくつかのCH3X化合物のメチル基化学シフトとX電気陰性度との関係は、X=methyl(Vollhardt&Schore,2014,p., 389)、従って不便なカーボン問題を避けます。ここでは、アルキル基が–I+R置換基として振る舞うことを示す。 このアプローチでは、いくつかの要因(場、立体または溶媒効果など)は暗黙的に無視されますが、現在利用可能な理論的および実験的証拠の多くは、したが
Cδ-Hδ+結合分極は、メタン(Lazzeretti、Zanasi、&Raynes、1987)について実験的に観察されており、水素に対する炭素のより大きな電気陰性度2.55対2と一貫している。,ポーリング-スケールの20(Allred、1961)。 このような分極パターンは、定量的な一致は通常modestえめであるが、添加モデルを通じて単純な炭化水素の双極子モーメント方向を予測することを可能にする(2-methylpropane:0.3D estimated vs.0.132D experimental)(Dean、1999)。
水素はインゴルドの置換基分類の標準として使用されているため(Krygowski&St??pie π,2005)では、アルキル基はa–I置換基(したがってa π電子吸引基)として分類されるべきである。 このような役割を図に示す。, 1どんな原子Yからメチル基へのπ結合分極についても、Yが炭素(例えば塩素)よりも電気陰性である場合には逆結合分極が期待される。
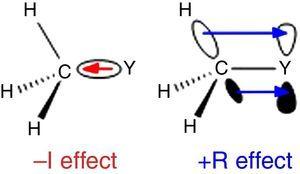
–i(左)および+R(右)原子Yに結合したメチル基の効果
図に示すように、アルキル基がsp2またはspハイブリダイズ原子に結合したとき、アルキルC-HまたはC–C π結合から連続した原子(最も単純なπ系)の空のp軌道に電子密度寄与によって異なる挙動が見られる。 1. したがって、p-メチル置換(McMahon&Kebarle、1977)によるフェノールおよび安息香酸の気相酸度の低下は、メチル置換基(実際にはメトキシ基よりも大きい)に対する有意なγドナー効果にのみ起因することができる。, しかし,孤立電子対がないため,アルキル基は非定型πドナー置換基と考えるべきである。 このようなπ結合/π系相互作用は、超共役相互作用における電子放出単位としてアルキル基のC-CまたはC-H結合(電子孤立対ではなく)が関与しているが、空のp軌道への孤立対を有する原子(例えば塩素)のπドナー挙動との類推によって容易に説明することができる(Mullins、2012)。, 興味深いことに、π→π*相互作用(負の超共役)は、電気陰性原子を欠くアルキル基については通常無視できる(Bocca、Pontes、&Basso、2004)。
いくつかの分子構造的特徴は、アルキル基の特性に基づいて合理化することができる。 例えば、メチルケトンに見られるより大きなCO結合長(acetone:exp. 1.210Å、calc。 1.193Å)関連するアルデヒドと比較して(アセトアルデヒド:exp. 1.209Å、calc。 1.,188Å)(Berry,Waltman,Pacansky,&Hagler,1995)は、両性イオン共鳴形態の安定化に起因する可能性がある(図参照)。 2)アルキル基γ-カルボニル炭素原子への寄付により、カルボニル基の二重結合の特徴を弱める。
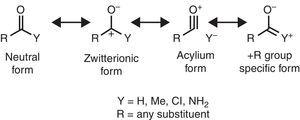
カルボニル化合物の中性(左)および両性イオン(右)共鳴形態。
超共役相互作用は、連続した原子Yのp軌道に対するC–H(またはC–C)結合の配置に依存し、ほぼ平行な配置に対応する最も効果的な相互作用である。 例えば、骨格面にほぼ垂直なトルエンCsp3–H結合は、他のCsp3–H結合よりもわずかに長い(0.002Å,Hameka&Jensen,1996)。, 超共役の幾何学的依存性は、プロペン(Liberles、O’Leary、Eilers、&Whitman、1972)またはアセトアルデヒド(Muñoz-Caro、Niño、&Moule、1994)のようなメチル置換不飽和化合物の立体配座分析を説明することを可能にする。
アルキル基のπドナー挙動のよく知られた結果として、アルキル置換はより多くの電子に富むアルケンおよびアレーンをもたらす(Libit&Hoffmann,1974)。, Sear反応におけるアルキル置換アレーンの高い反応性は,π電子寄与による対応するホエランド中間体の安定化に起因することが分かった。
アルキル基の–I+R挙動により、以下に示すように、双極子モーメント、分光特性および反応性(気相および溶液媒体中)などのアルキル置換化合物の多くの特徴を説明することができる。
双極子モーメント
脂肪族化合物中のアルキル基の電子吸引挙動も双極子モーメントに反映される。, したがって、プロパンおよび2-メチルプロパンの双極子モーメントベクトル(Tasi et al.,1997)、ならびにいくつかの置換ビシクロオクタン(Böhm&Exner,2004)は、水素と比較してメチル基の吸引効果(–I)に起因する可能性がある(図参照)。 3).
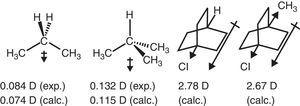
プロパンおよび置換ビシクロオクタンの双極子モーメント。
対照的に、ニトロベンゼンとベンゾニトリルの双極子モーメントのp-メチル置換による上昇を説明するためには、メチル基(+R)のπドナー特性が必要である(Brown,1959)。 4).
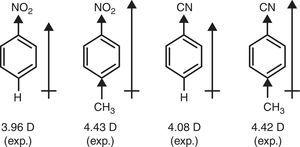
ベンゼン誘導体の双極子モーメント。
分子双極子モーメントは、現在の計算方法によって確実に計算することができます。, 興味深いことに、単純な炭化水素のセットのための計算された双極子モーメントベクトル(Tasi et al.,1997)は、メチル基の二重の役割を推論することを可能にした:sp3炭素原子に結合したときに電子吸引性が、sp2またはsp3炭素に結合したときに電子
アルキル置換基のこのような二重挙動は、ヘテロ原子を有する化合物についても観察される。 したがって、アンモニア(NH3、1.47D;MeNH2、1.31D;Me2NH、1.01D;Me3N、0)上の連続したメチル置換については、徐々に双極子モーメントの減少が観察される。,61D)(Le Fèvre&Russell,1947),窒素電子密度の漸進的減少と一致して(Hehre&Pople,1970). 対照的に、双極子モーメント強化(1.53Dから1.68D)(Nelson、Lide、&Maryott、1967)は、アニリン上のN、N-ジメチル置換(Targema、Obi-Egbedi、&Adeoye、2013)、アミノ基のγドナー文字の上昇と一貫して(Hinchliffe&kidd,1980)メチル置換基の+R寄与による。,
分光学的性質
多くの有機化合物の分光学的性質は、一般的な特徴としてアルキル基の–I+R挙動を仮定することによって容易に合理化することができる。 したがって、原子のNMR化学シフトは、対応する核位置における電子密度の実験的尺度とみなすことができるが、異方性磁場のような他の効果も関与することができる。 Sp3炭素原子上のメチル置換基によって誘導されるダウンフィールドシフト(+9.6ppmで13C NMR)または対応する結合した水素原子(+0。,63ppm in1H NMR)(Pretch,Bühlmann,&Badertscher,2009)は、典型的な–I基(ハロゲン原子など)の挙動と一致している。
アルケンのNMR化学シフトに対するアルキル置換効果は、α位の電子密度の減少(+12.9ppmで13C NMR、+0.45ppmで1H NMR)、β位の密度の上昇(-7.4ppmで13C、-0.31/-0.40ppmで1H)を示し、a–I+R効果と一貫しているが、異方性効果(環電流など)も役割を果たす可能性がある。 このようなa–I+R挙動は、13C NMR分光法(+8.,Αの位置のための5ppm、βの位置のための-3.6ppm)。
γ-系におけるアルキル置換基の二分的挙動(α原子に対する電子密度の上昇,β原子に対する電子密度の減少)は,単純な挙動(a+i効果など)に基づいて説明することはできない。
A-I+R挙動(Meier、2007)は、窒素ハイブリダイゼーション(脂肪族アミンのダウンフィールドシフト、アニリンやアミドなどのNsp2ベアリング化合物のアップフィールドシフト)に応じて、アミンとアミド上のアルキル置換のための15N NMR分光法によって観察される。,
NMR結合定数は、置換基の電子的性質(ならびにいくつかの幾何学的特徴)にも依存する。 したがって、有意な減少は、メチル置換(trans、-2.3Hz;cis、-1.6Hz;gem、-0.4Hz)、フッ素原子(trans、-6.3Hz;cis、-6.9Hz;gem、-5.7Hz)のような典型的な電子吸引基からのデータと定性的な一致で、1H–1Hカップリング定数のために見出される。 脂肪族化合物の13C-1Hカップリング定数に対するメチル置換のための正の寄与(+1。,0Hz)、また、他の–I基(フッ素、+24Hz)からのものと定性的に一致している。
赤外分光法は、対応する置換基Yの関数としてのカルボニル化合物のCO延伸周波数によって示されるように、置換基特性にも敏感であり、これは共 2). 脂肪族アルデヒドを取ることによって(ca., 1725cm-1)参考として、a+I置換基(acetyltrimethylsilane,1645cm–1:Soderquist&Hsu,1982)によって誘導される赤方偏移(波数減少)は、両性イオン型の安定化に起因する可能性がある。 代わりに、a–I置換基によって引き起こされるブルーシフト(acyl chlorides,>1800cm–1:Pretsch et al.,2009)は、二つの代替または同時メカニズム(両性イオン形態の不安定化および/またはアシリウムイオン担持形態の寄与)によって説明することができる。 最後に、+R置換基によって引き起こされる赤方偏移(amides,ca., 1680cm-1:Pretsch et al.,2009)は、特定の共鳴形態の寄与に起因する可能性がある。 アルキル基によって誘導されるわずかな赤方偏移(methylketones,ca. 1715cm-1)は正味の電子供与効果を示す(したがって、+R効果が–I特性に対して優勢である)。 カルボニル結合アルキル基の正味ドナー効果は、ホルムアルデヒド(2.88D)に対するアセトン(2.33D)の大きな双極子モーメントと一致している(Nelson et al., 1967).
多くの化合物のUV–Visスペクトルに及ぼすアルキル基の影響は、電子効果によって説明することができる。, したがって、α、β-不飽和化合物(α位で+10nm、β位で+12nm)、共役ポリエン(+5nm)またはベンゼン誘導体(+3.0nm)の紫外線吸収バンド上のアルキル基によって誘導されるバクロミックシフトは、典型的なγ-ドナー基(例えば、塩素)の効果と定性的に一致している。
気相酸-塩基反応性
aqueous液中の脂肪族アミンの相対塩基性は、アルキル基の仮定された+I効果に起因している(Sorrell、2006)。, Interestingly, the irregular basicity order of amines in water (Me2NH>MeNH2>Me3N>NH3, as shown by the pKa values for the corresponding conjugated acids: 10.77>10.62>9.80>9.,246)(Dean,1999)は、気相中のアミンの系統的塩基度秩序によって示されるように、溶媒効果によって汚染されている(Me3N>Me2NH>MeNH2>NH3)(Brauman,Riveros,&Blair,1971)。, 気相塩基度秩序は、通常、想定される+Iアルキル効果(Carter、2007)に起因することができるが、メチル置換による窒素電子密度の減少は、分子静電ポテンシャル計算(Baeten、De Proft、&Geerlings、1995)によって確かに観測されており、メチル基の–I挙動を示している。, 実際には、脂肪族アミンの気相塩基度秩序は、アルキル基分極性による置換アンモニウムイオンの安定化の増加に起因するはずである(Aue、Webb、&Bowers、1976)。
aqueous液中のアルコールの相対酸度(H2O>MeOH>EtOH>iPrOH>tBuOH)も、いくつかの教科書では、アルキル+I効果が想定されている(Johnson,1999;Solomons,fryhle,&snyder,2016)., 逆の酸性度は気相で見られるので、水中のアルコールの相対的な酸性度は、より大きなアルコキシドアニオンの溶媒和エンタルピーの低い大きさに起因するはずである(Brauman&Blair,1969)。
アルキル基の電子特性に関する議論は、カルバニオンにも適用することができる。 したがって、単純なカルバニオン(メチル>エチル>イソプロピル>tert-ブチル)の”教科書”安定度秩序は、アルキル基の仮定された+I誘導効果に起因している(Burrows et al.,,2013;Chaloner,2015;Roos&Roos,2014;Smith,2013). しかし、気相カルバニオン安定性(tBu>Me>iPr>Et)については、二つの対向アルキル効果(DePuy et al.,1989):アルキル分極性(すなわち、n→π*超共役)と不安定化傾向(炭素孤立電子対のp様挙動を仮定することにより、a+Rの役割と一貫して)による安定化機構。,
他の反応中間体の安定性は、アルキル基効果に基づいて評価することもできる。 したがって、カルボカチオンのよく知られた安定秩序(三次>二次>一次>メチル)は、正の誘導効果に起因することがある(Chaloner、2015;Roos&Roos、2014)。, 興味深いことに、超共役は、カルボカチオンの安定次数の代替説明として多くの教科書に提示されている(Brown,Iverson,Anslyn,&Foote,2013;Burrows et al.,2013)通常のあいまいな書き込みは、両方の説明が同じ現象の二つの異なる記述または同じ方向に遊んでいる二つの同時メカニズムのいずれかに対応, いずれにせよ、カルボカチオンの安定秩序は、他の相互作用(アルキル分極性など)も関与しているが、超共役(したがって、最も単純なπ系である空いているp軌道上のa+R挙動)に起因するはずである(Aue、2011)。
フリーラジカルはカルボカチオンと同じ安定秩序を示し,アルキル置換による安定化を示した。 このような安定性秩序は、仮定された+I挙動に基づいて正当化されるかもしれないが、+R効果は、孤立電子対を有する原子によるフリーラジカルの安定化(Zipse、2006)と同様に、代わりに見なすことができる(Zipse、2006)。,
溶液中の反応性
aqueous液中の単純なカルボン酸の相対酸度(酢酸>プロピオン酸>酪酸)は、アルキル基の仮定された+I効果を説明するために、いくつかの教科書で使用されている(Sorrell、2006)。 興味深いことに、エンタルピーが代わりにみなされると逆の順序が見つかり(Christensen,Izatt,&Hansen,1967)、aqueous液中の酸性度の順序は水和エントロピーに起因するはずであることを示している。, したがって、液体水の有意な格子次数(蒸発エントロピーは118.89Jmol–1K–1に等しく、caの典型的な値とは対照的である。 88Jmol-1K-1ほとんどの液体については、Dean、1999)は、反応エネルギー論にかなりの変化をもたらすことができる。 特に、無極性分子(または部分)の水和は、さらなる溶媒格子秩序をもたらす(Blokzijl&Engberts、1993)。 結果として、aqueous液中の実験データからのアルキル基誘導効果は、しばしば水和エントロピーによってマスクされる(Calder&Barton、1971)。, 気相中の単純カルボン酸の相対酸度(Yamdagni&Kebarle,1973)およびアセトニトリル(Eckert et al.,2009)は、水分補給エントロピーが果たす主要な役割と一致している。
酢酸と比較してピバル酸の酸性度が低く、通常はアルキル基の+I効果に起因する(Smith、2008)が、反応エンタルピーを考慮すると逆転する(Eckert et al., 2009).,aqueous液中の単純カルボン酸の酸性度に対する仮定された+iアルキル基効果は,溶媒効果に由来するアーチファクトに起因すると考えられる。 中性溶質の体積の増加は水和エントロピーの上昇をもたらすのに対し、イオン種については逆の関係が見られる(Graziano、2009)。 結果として、アルキル置換(分子体積の増加による)は、水中の非イオン化酸の安定化(ギブス自由エネルギー項で)および対応するカルボキシレートアニオンの不安定化をもたらし、酸性度の低下をもたらす。,
aqueous液中の酢酸と比較してギ酸の酸性度が大きい(pka値:それぞれ3.751および4.756、Dean、1999)も、誘導効果の適用の例として多くの教科書で議論されている(Hart,Hadad,Craine,&Hart,2012;Hornback,2006;Okuyama&Maskill,2014;Roos&roos,2014). 非常に類似した反応エンタルピーがギ酸および酢酸の解離反応に関与するので(Christensen et al.,,1967)、ギ酸のより大きな酸性度は、実際に水和エントロピーの違いに起因しなければならない。
結論
誘導効果と共鳴効果を明確に理解することは、有機化学の健全な学習のための主要な鍵である(Mullins、2008)。 驚くべきことに、ほとんどユビキタスなアルキル基は、γ-ドナー(+I)基として多くの教科書で誤って提示されています。 しかし,隣接原子のハイブリダイゼーションに依存するアルキル置換基によって二重挙動を示した。, したがって,脂肪族鎖に結合したアルキル基はγ-アクセプタ(–i,水素に対する炭素の大きな電気陰性度と一貫して)として振る舞い,γ-系に結合したアルキル基はγ-ドナーとして作用する(超共役相互作用による+R)。 多くの実験および理論データ(双極子モーメント,NMR,IRおよびUVスペクトル,反応性)はこのような二重挙動と一致した。,
ここで検討したすべてのデータの全体分析により、有機化合物の分光学的特性および反応性特性に関するすべての議論において有効な特徴として、アルキル基に対する小さな–I効果および有意な+R挙動を推測することができる。
利益相反
著者は利益相反を宣言していません。