Introduzione
Gli effetti sostituenti costituiscono un concetto chiave per la comprensione della reattività e del comportamento spettroscopico dei composti organici (Krygowski& St??pień, 2005). In un approccio semplice, gli effetti sostituenti possono essere classificati in base al meccanismo di interazione con il centro reattivo come effetti induttivi (attraverso i legami σ) o di risonanza (attraverso i legami π)., Tuttavia, alcuni ulteriori termini (come sterico, campo o solvente effetti) sarebbero necessari per una descrizione approfondita degli effetti sostituenti.
Dal momento che il Ingold classificazione di elettronica sostituente effetti (Ingold, 1953), il gruppo alchile è stato considerato come una σ-donatore sostituente (+i, Ingold nomenclatura), nella maggior parte dei libri di testo di Chimica Organica (Burrows, Holman, Parsons, Pilling, & Prezzo, 2013; Hornback, 2006; Roos & Roos, 2014; Smith, 2013; Vollhardt & Schore, 2014)., Tuttavia, le critiche dell’Eğe a un punto di vista così semplicistico dovrebbero essere rimarcate:
“In acqua, l’acido propanoico è leggermente più debole dell’acido acetico. La natura dell’effetto induttivo di un gruppo alchilico è discussa dai chimici. Gruppi alchilici stabilizzano carbocations e in quel ruolo sembrano essere rilascio di elettroni. Aumentano anche la basicità delle ammine, suggerendo ancora una volta che stanno rilasciando elettroni. D’altra parte, sebbene l’alcol tert-butilico (pKa 19) sia un acido più debole dell’etanolo (pKa 17) in acqua, è acido più forte nella fase gassosa., Questa osservazione sperimentale suggerisce che i gruppi alchilici possono stabilizzare anioni e cationi e che la solvatazione svolge un ruolo importante nel determinare le acidità relative. Pertanto è necessaria una parola di cautela. Le acidità relative su cui si basano le generalizzazioni presentate in questo capitolo sono state determinate in acqua. Nella fase gassosa, si osservano spesso inversioni nell’ordine dei composti correlati.”(Eğe, 1999, p. 107)
Alcuni effetti di sostituzione alchilica sono stati spesso spiegati nei libri di testo in modi contraddittori o enigmatici., Pertanto, le differenze di spostamento chimico tra i gruppi CH3 e CH2 sono attribuite nel libro di Hornback al fatto che” il carbonio è leggermente più elettronegativo dell’idrogeno ” (Hornback, 2006, p. 549) nonostante il gruppo alchilico sia stato precedentemente classificato come un debole sostituente induttivo che dona elettroni (Hornback, 2006, p. 117). Nel libro di testo di Vollhardt, la relazione tra i cambiamenti chimici del gruppo metilico per un certo numero di composti CH3X e l’elettronegatività X è illustrata da una tabella priva di una voce per X=metile (Vollhardt & Schore, 2014, p., 389), evitando così lo scomodo problema del carbonio.
Mostro qui che il gruppo alchilico si comporta come sostituente a-I + R. Sebbene alcuni fattori (come gli effetti di campo, sterici o solventi) siano implicitamente ignorati in questo approccio, molte evidenze teoriche e sperimentali attualmente disponibili possono quindi essere descritte in modo semplice.
Una polarizzazione del legame Cδ H Hδ+ è stata osservata sperimentalmente per il metano (Lazzeretti, Zanasi,& Raynes, 1987), coerentemente con la maggiore elettronegatività del carbonio rispetto all’idrogeno, 2.55 vs. 2.,20 nella scala di Pauling (Allred, 1961). Tale modello di polarizzazione consente di prevedere la direzione del momento di dipolo di idrocarburi semplici attraverso modelli additivi, sebbene l’accordo quantitativo sia solitamente modesto (2-metilpropano: 0.3 D stimato vs. 0.132 D sperimentale) (Dean, 1999).
Poiché l’idrogeno è usato come standard nella classificazione dei sostituenti di Ingold (Krygowski& St??pień, 2005), il gruppo alchilico dovrebbe essere classificato come sostituente a –I (quindi un gruppo di ritiro degli elettroni σ). Tale ruolo è illustrato in Fig., 1 per la polarizzazione del legame σ da qualsiasi atomo Y a un gruppo metilico, anche se la polarizzazione del legame inverso è prevista quando Y è più elettronegativo del carbonio (ad esempio, cloro).
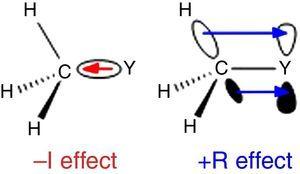
–I (sinistra) e +R (destra) effetti di un gruppo metilico legato ad un atomo Y.
Un comportamento diverso si trova per i gruppi alchilici quando sono collegati a sp2 o sp-atomi ibridati a causa della donazione di densità elettronica da legami alchilici C–H o C–C σ all’orbitale p vuoto dell’atomo contiguo (il più semplice sistema π), come mostrato in Fig. 1. Pertanto, la diminuzione dell’acidità in fase gassosa per il fenolo e l’acido benzoico attraverso la sostituzione p-metile (McMahon & Kebarle, 1977) può essere attribuita solo a un significativo effetto π-donatore per il sostituente metilico (anzi, più grande di quello per il gruppo metossi)., Tuttavia, il gruppo alchilico dovrebbe essere considerato come un sostituente atipico π-donatore a causa della mancanza di coppie di elettroni solitari. Tale interazione σ-legame / π-sistema, denominata iperconiugazione (Mullins, 2012) può essere facilmente spiegata per analogia con il comportamento π-donatore di un atomo a coppia solitaria (ad esempio, cloro) a un orbitale p vuoto, sebbene legami C-C o C–H (piuttosto che coppie di elettroni solitari) del gruppo alchilico siano coinvolti come unità di rilascio di elettroni nelle interazioni iperconiugative., È interessante notare che le interazioni π→σ* (iperconiugazione negativa) sono solitamente trascurabili per i gruppi alchilici privi di atomi elettronegativi (Bocca, Pontes, & Basso, 2004).
Alcune caratteristiche strutturali molecolari possono essere razionalizzate sulla base delle proprietà del gruppo alchilico. Ad esempio, le lunghezze di legame CO più grandi trovate nei metilchetoni (acetone: exp. 1.210 Å, calc. 1.193 Å) rispetto alle aldeidi correlate (acetaldeide: scad. 1.209 Å, calc. 1.,188Å) (Berry, Waltman, Pacansky, & Hagler, 1995) può essere attribuito alla stabilizzazione della forma di risonanza zwitterionica (vedi Fig. 2) attraverso il gruppo alchilico π-donazione all’atomo di carbonio carbonilico, indebolendo così la caratteristica del doppio legame del gruppo carbonilico.
Forme di risonanza neutra (sinistra) e zwitterionica (destra) di un composto carbonilico.
Le interazioni iperconiugative dipendono dalla disposizione dei legami C–H (o C–C) rispetto all’orbitale p dell’atomo contiguo Y, l’interazione più efficace corrispondente a una disposizione quasi parallela. Ad esempio, il legame Csp3–H di toluene quasi perpendicolare al piano della struttura è leggermente più lungo degli altri legami Csp3–H (di 0,002 Å, Hameka & Jensen, 1996)., La dipendenza geometrica dell’iperconiugazione consente di spiegare l’analisi conformazionale di composti insaturi metil-sostituiti, come propene (Liberles, O’Leary, Eilers, & Whitman, 1972) o acetaldeide (Muñoz-Caro, Niño, & Moule, 1994).
Come conseguenza ben nota del comportamento del donatore π del gruppo alchilico, la sostituzione alchilica produce più alcheni e areni ricchi di elettroni (Libit & Hoffmann, 1974)., L’elevata reattività di un arene alchil-sostituito in una reazione di scottatura può quindi essere attribuito alla stabilizzazione del corrispondente Wheland intermedio attraverso la donazione π-elettrone.
Il comportamento –I+R del gruppo alchilico consente di spiegare una serie di caratteristiche dei composti alchil-sostituiti, come i momenti di dipolo, le proprietà spettroscopiche e la reattività (in fase gassosa e mezzi di soluzione), come mostrato di seguito.
Momenti di dipolo
Il comportamento di ritiro degli elettroni del gruppo alchilico nei composti alifatici si riflette anche nei momenti di dipolo., Quindi, i vettori del momento di dipolo per propano e 2-metilpropano (Tasi et al., 1997), così come alcuni bicicloottani sostituiti (Böhm & Exner, 2004) possono essere attribuiti all’effetto di ritiro (–I) del gruppo metilico rispetto all’idrogeno (vedi Fig. 3).
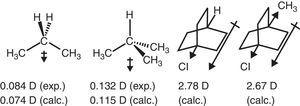
Momenti di dipolo di propano e bicicloottani sostituiti.
Al contrario, il carattere π-donatore del gruppo metilico (+R) è richiesto per spiegare l’aumento dei momenti dipolici di nitrobenzene e benzonitrile attraverso la sostituzione p-metile (Brown, 1959) (vedi Fig. 4).
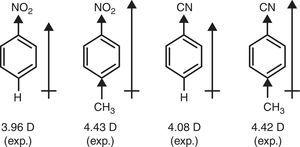
Momenti di dipolo dei derivati del benzene.
I momenti di dipolo molecolare possono essere calcolati in modo affidabile con gli attuali metodi computazionali., È interessante notare che i vettori del momento di dipolo calcolati per un insieme di idrocarburi semplici (Tasi et al., 1997) hanno permesso di dedurre un duplice ruolo per il gruppo metilico: ritiro di elettroni quando collegato agli atomi di carbonio sp3, ma donazione di elettroni quando legato ai carboni sp2 o sp3.
Un tale doppio comportamento del sostituente alchilico è anche osservato per i composti portatori di eteroatomee. Pertanto, si osserva una graduale diminuzione del momento di dipolo per la successiva sostituzione di metile sull’ammoniaca (NH3, 1,47 D; MeNH2, 1,31 D; Me2NH, 1,01 D; Me3N, 0.,61 D) (Le Fèvre & Russell, 1947), in accordo con la progressiva diminuzione della densità elettronica dell’azoto (Hehre & Pople, 1970). Al contrario, un momento di dipolo di miglioramento (da 1.53 D a 1,68 D) (Nelson, Lide, & Maryott, 1967) è stato trovato per N,N-dimetil sostituzione anilina (Targema, Obi-Egbedi, & Adeoye, 2013), coerentemente con la crescita della π-donatore di carattere per il gruppo amminico (Hinchliffe & Kidd, 1980), a causa +R contributi di metile sostituenti.,
Proprietà spettroscopiche
Le proprietà spettroscopiche di molti composti organici possono essere facilmente razionalizzate assumendo il comportamento a –I+R per il gruppo alchilico come caratteristica generale. Pertanto, lo spostamento chimico NMR di un atomo può essere considerato come una misura sperimentale della densità elettronica nella posizione del nucleo corrispondente, sebbene possano essere coinvolti anche altri effetti, come i campi magnetici anisotropici. Turni di downfield indotti da un sostituente metilico su atomi di carbonio sp3 (+9,6 ppm in 13C NMR) o sui corrispondenti atomi di idrogeno legati (+0.,63ppm in 1H NMR) (Pretsch, Bühlmann, & Badertscher, 2009) sono coerenti con il comportamento dei gruppi tipici –I (come gli atomi di alogeno).
Gli effetti di sostituzione alchilica sugli spostamenti chimici NMR degli alcheni mostrano una diminuzione della densità elettronica in posizione α (+12,9 ppm per 13C NMR; +0,45 ppm per 1H NMR), così come un aumento della densità in posizione β (-7,4 ppm per 13C; -0,31 / -0,40 ppm per 1H), coerentemente con l’effetto a –I+R, sebbene anche gli effetti anisotropici (come le correnti ad anello) possano svolgere un ruolo. Tale comportamento a-I + R si trova anche per gli alchini, secondo la spettroscopia 13C NMR (+8.,5ppm per la posizione α, -3.6 ppm per la posizione β).
Il comportamento dicotomico dei sostituenti alchilici sui sistemi π (aumento della densità elettronica per atomo α, diminuzione della densità elettronica per atomo β) non può essere spiegato sulla base di un comportamento semplice (come l’effetto a +I).
Il comportamento A –I+R (Meier, 2007) è osservato attraverso la spettroscopia 15N NMR per la sostituzione alchilica su ammine e ammidi a seconda dell’ibridazione dell’azoto (turni di downfield per ammine alifatiche, turni di upfield per composti portatori di Nsp2-come aniline e ammidi).,
Le costanti di accoppiamento NMR dipendono anche dalle proprietà elettroniche sostituenti (così come da alcune caratteristiche geometriche). Pertanto, si riscontra una diminuzione significativa per le costanti di accoppiamento 1H–1H attraverso la sostituzione di metile (trans, -2,3 Hz; cis, -1,6 Hz; gem, -0,4 Hz), in accordo qualitativo con i dati dei tipici gruppi di ritiro degli elettroni, come l’atomo di fluoro (trans, -6,3 Hz; cis, -6,9 Hz; gem, -5,7 Hz). Il contributo positivo per la metil-sostituzione sulle costanti di accoppiamento 13C-1H dei composti alifatici (+1.,0Hz), è anche qualitativamente coerente con quelli di altri gruppi-I (fluoro, +24Hz).
La spettroscopia infrarossa è anche sensibile alle proprietà sostituenti, come illustrato dalla frequenza di CO stretching dei composti carbonilici in funzione del corrispondente sostituente Y, che può essere razionalizzato in termini di forme di risonanza (Fig. 2). Prendendo un’aldeide alifatica (ca., 1725cm–1) come riferimento, il redshift (diminuzione del numero d’onda) indotto da un sostituente +I (acetiltrimetilsilano, 1645cm–1: Soderquist & Hsu, 1982) può essere attribuito alla stabilizzazione della forma zwitterionica. Invece, il blueshift provocato da a-I sostituente (cloruri acilici, >1800cm-1: Pretsch et al., 2009) può essere spiegato per mezzo di due meccanismi alternativi o simultanei (destabilizzazione della forma zwitterionica e/o contributo di una forma portatrice di ioni acili). Infine, i redshifts provocati da + R sostituenti (ammidi, ca., 1680cm-1: Pretsch et al., 2009) può essere attribuito al contributo di una specifica forma di risonanza. Il leggero redshift indotto dal gruppo alchilico (metilchetoni, ca. 1715cm-1) mostra un effetto di donazione di elettroni netto (quindi, una predominanza dell’effetto +R rispetto alle proprietà –I). L’effetto donatore netto del gruppo alchilico legato al carbonile è coerente con il momento di dipolo più grande dell’acetone (2.88 D) rispetto alla formaldeide (2.33 D) (Nelson et al., 1967).
L’influenza del gruppo alchilico sugli spettri UV–Vis di molti composti può anche essere spiegata in termini di effetti elettronici., Pertanto, gli spostamenti batocromici indotti da gruppi alchilici su bande di assorbimento UV di α, composti β-insaturi (+10 nm in posizione α, +12 nm in posizione β), polieni coniugati (+5 nm) o derivati del benzene (+3,0 nm) sono qualitativamente coerenti con gli effetti dei tipici gruppi π-donatori (ad esempio, cloro).
Fase gassosa reattività acido–base
Le basi relative delle ammine alifatiche in soluzione acquosa sono state attribuite al presunto effetto +I del gruppo alchilico (Sorrell, 2006)., Interestingly, the irregular basicity order of amines in water (Me2NH>MeNH2>Me3N>NH3, as shown by the pKa values for the corresponding conjugated acids: 10.77>10.62>9.80>9.,246) (Dean, 1999), è contaminato da solventi effetti, come illustrato dalla sistematica ordine di basicità delle ammine in fase gas (Me3N>Me2NH>MeNH2>NH3) (Brauman, Riveros, & Blair, 1971)., Sebbene l’ordine di basicità della fase gassosa possa essere attribuito all’effetto alchilico +I solitamente assunto (Carter, 2007), una diminuzione della densità elettronica dell’azoto attraverso la sostituzione metile è stata effettivamente osservata mediante calcoli di potenziale elettrostatico molecolare (Baeten, De Proft, & Geerlings, 1995), indicando così il comportamento a –I per il gruppo metilico., In realtà, l’ordine di basicità della fase gassosa delle ammine alifatiche dovrebbe essere attribuito alla crescente stabilizzazione degli ioni ammonio sostituiti a causa della polarizzabilità del gruppo alchilico (Aue, Webb, & Bowers, 1976).
in relazione acidities di alcoli in soluzione acquosa (H2O>MeOH>EtOH>iPrOH>tBuOH) sono state attribuite in alcuni libri di testo per la presunta alchil +I effetto (Johnson, 1999; Solomons, Fryhle, & Snyder, 2016)., Poiché l’ordine di acidità inverso si trova in fase gassosa, le acidità relative degli alcoli nell’acqua dovrebbero essere attribuite alle magnitudini inferiori delle entalpie di solvatazione per anioni alcossido più grandi (Brauman & Blair, 1969).
La discussione sulle proprietà elettroniche dei gruppi alchilici può essere applicata anche ai carbanioni. Pertanto, l’ordine di stabilità “da manuale” per carbanioni semplici (metil>etile>isopropile>tert-butile) è stato attribuito all’effetto induttivo assunto +I dei gruppi alchilici (Burrows et al., nel 2013, Chaloner, 2015, Roos & Roos, 2014; Smith, 2013). Tuttavia, si trova un ordine irregolare per le stabilità dei carbanioni in fase gassosa (tBu>Me>iPr>Et), in accordo con la concomitanza di due effetti alchilici opposti (DePuy et al., 1989): un meccanismo stabilizzante attraverso la polarizzabilità alchilica (cioè, n→σ* iperconiugazione) e una tendenza destabilizzante (coerentemente con il ruolo di a +R, assumendo un comportamento simile a p per la coppia solitaria di carbonio).,
La stabilità di altri intermedi di reazione può anche essere valutata sulla base degli effetti del gruppo alchilico. Pertanto, il noto ordine di stabilità per le carbocazioni (terziario>secondario>primario>metile) è stato talvolta attribuito ad un effetto induttivo positivo (Chaloner, 2015; Roos & Roos, 2014)., È interessante notare che l’iperconiugazione è presentata in molti libri di testo come una spiegazione alternativa per l’ordine di stabilità delle carbocazioni (Brown, Brownon, Anslyn, & Foote, 2013; Burrows et al., 2013) sebbene la solita scrittura ambigua impedisca di accertare se entrambe le spiegazioni corrispondano a due diverse descrizioni dello stesso fenomeno o a due meccanismi simultanei che giocano nella stessa direzione., Ad ogni modo, l’ordine di stabilità per le carbocazioni dovrebbe essere attribuito all’iperconiugazione (quindi, comportamento a+R su un orbitale p vacante, il sistema π più semplice), sebbene siano coinvolte anche altre interazioni (come la polarizzabilità alchilica) (Aue, 2011).
I radicali liberi mostrano lo stesso ordine di stabilità dei carbocati, indicando così la stabilizzazione attraverso la sostituzione alchilica. Sebbene un tale ordine di stabilità possa essere giustificato sulla base di un presunto comportamento + I, l’effetto + R può essere alternativamente considerato, analogamente alla stabilizzazione dei radicali liberi da parte di atomi portatori di coppie solitarie (Zipse, 2006).,
Reattività in soluzione
Le acidità relative degli acidi carbossilici semplici in soluzione acquosa (acido acetico>acido propionico>acido butirrico) sono state utilizzate in alcuni libri di testo per illustrare l’effetto assunto +I del gruppo alchilico (Sorrell, 2006). È interessante notare che l’ordine inverso si trova quando si considerano invece le entalpie (Christensen, Izatt, & Hansen, 1967), indicando così che l’ordine di acidità in soluzione acquosa dovrebbe essere attribuito alle entropie di idratazione., Pertanto, l’ordine reticolare significativo dell’acqua liquida (entropia di vaporizzazione pari a 118,89 Jmol–1K–1, in contrasto con i valori tipici di ca. 88Jmol–1K-1 per la maggior parte dei liquidi, Dean, 1999) può introdurre notevoli cambiamenti sull’energetica di reazione. In particolare, l’idratazione delle molecole apolari (o parti) porta ad un ulteriore ordinamento del reticolo solvente (Blokzijl & Engberts, 1993). Di conseguenza, gli effetti induttivi del gruppo alchilico dai dati sperimentali in soluzione acquosa sono spesso mascherati da entropie di idratazione (Calder & Barton, 1971)., Acidità relative degli acidi carbossilici semplici in fase gassosa (Yamdagni & Kebarle, 1973 )e acetonitrile (Eckert et al., 2009) sono coerenti con il ruolo principale svolto dalle entropie di idratazione.
La minore acidità dell’acido pivalico rispetto all’acido acetico, solitamente attribuita all’effetto +I assunto del gruppo alchilico (Smith, 2008), viene invertita quando vengono considerate le entalpie di reazione (Eckert et al., 2009).,
Il presunto effetto del gruppo alchilico +I sull’acidità degli acidi carbossilici semplici in soluzione acquosa può quindi essere attribuito a un artefatto derivato dagli effetti del solvente. Mentre un aumento di volume di soluti neutri porta ad un aumento dell’entropia di idratazione, la relazione inversa si trova per le specie ioniche (Graziano, 2009). Di conseguenza, la sostituzione alchilica (attraverso un aumento del volume molecolare) porta alla stabilizzazione (in termini di energia libera di Gibbs) dell’acido non ionizzato in acqua e alla destabilizzazione del corrispondente anione carbossilato, con conseguente diminuzione dell’acidità.,
Il più grande acidità dell’acido formico in confronto con l’acido acetico in soluzione acquosa (valori di pKa: 3.751 e 4.756, rispettivamente, Dean, 1999) è stato anche discusso in molti libri di testo, come un esempio di applicazione di effetti induttivi (Hart, Hadad, Craine, & Hart, 2012; Hornback, 2006; Okuyama & Maskill, 2014; Roos & Roos, 2014). Poiché le entalpie di reazione molto simili sono coinvolte nelle reazioni di dissociazione degli acidi formici e acetici (Christensen et al.,, 1967), la maggiore acidità dell’acido formico deve essere effettivamente attribuita alle differenze di entropia di idratazione.
Conclusioni
Una chiara comprensione degli effetti induttivi e di risonanza è una chiave importante per un sano apprendimento della chimica organica (Mullins, 2008). Sorprendentemente, il gruppo alchilico quasi onnipresente è stato erroneamente presentato in molti libri di testo come gruppo σ-donatore (+I). Tuttavia, un doppio comportamento è mostrato dai sostituenti alchilici a seconda dell’ibridazione dell’atomo vicino., Pertanto, i gruppi alchilici legati alle catene alifatiche si comportano come σ-accettori (–I, coerentemente con la maggiore elettronegatività del carbonio rispetto all’idrogeno), mentre quelli collegati ai sistemi π agiscono come π-donatori (+R, a causa di interazioni iperconiugative). Un certo numero di dati sperimentali e teorici (momenti di dipolo, spettri NMR, IR e UV, reattività) concordano con un comportamento così duplice.,
L’intera analisi di tutti i dati qui considerati consente di dedurre un piccolo effetto –I e un significativo comportamento +R per il gruppo alchilico come caratteristica valida in tutte le discussioni sulle proprietà spettroscopiche e di reattività dei composti organici.
Conflitto di interessi
L’autore non dichiara alcun conflitto di interessi.