Bowtie indiziert das Referenzgenom anhand eines Schemas, das auf der Burrows-Wheeler-Transformation (BWT) und dem FM-Index basiert . Ein Bowtie-Index für das menschliche Genom passt in 2.2 GB auf der Festplatte und hat einen Speicherbedarf von jeweils nur 1.3 GB, sodass er auf einer Workstation mit weniger als 2 GB RAM abgefragt werden kann.,
Die gebräuchliche Methode zur Suche in einem FM-Index ist der exakt übereinstimmende Algorithmus von Ferragina und Manzini . Bowtie übernimmt diesen Algorithmus nicht einfach, da die genaue Übereinstimmung keine Sequenzierungsfehler oder genetischen Variationen zulässt. Wir führen zwei neuartige Erweiterungen ein, die die Technik für die Ausrichtung kurzer Lesevorgänge anwendbar machen: einen qualitätsbewussten Backtracking-Algorithmus, der Fehlanpassungen zulässt und qualitativ hochwertige Ausrichtungen begünstigt.und „doppelte Indizierung“, eine Strategie, um übermäßiges Backtracking zu vermeiden., Der Bowtie Aligner verfolgt eine ähnliche Politik wie Maq ‚ s, da er eine kleine Anzahl von Fehlanpassungen innerhalb des hochwertigen Endes jedes Lesevorgangs zulässt und die Summe der Qualitätswerte an nicht übereinstimmenden Ausrichtungspositionen an eine Obergrenze setzt.
Burrows-Wheeler Indizierung
Die BWT ist eine reversible Permutation der Zeichen in einem Text. Obwohl ursprünglich im Rahmen der Datenkomprimierung entwickelt, ermöglicht die BWT-basierte Indizierung die effiziente Suche großer Texte in geringem Speicherbedarf., Es wurde auf Bioinformatik-Anwendungen angewendet, einschließlich Oligomerzählung , Ausrichtung des gesamten Genoms , Tiling Microarray-Sondendesign und Smith-Waterman-Ausrichtung auf eine Referenz in menschlicher Größe .
Die Burrows-Wheeler-Transformation eines Textes T, BWT (T), ist wie folgt aufgebaut. Das Zeichen $ wird an T angehängt, wobei $ nicht in T und lexikographisch kleiner ist als alle Zeichen in T. Die Burrows-Wheeler-Matrix von T wird als Matrix konstruiert, deren Zeilen alle zyklischen Rotationen von T$umfassen. Die Zeilen werden dann lexikographisch sortiert., BWT (T) ist die Folge von Zeichen in der Spalte ganz rechts in der Burrows-Wheeler-Matrix (Abbildung 1a). BWT(T) hat die gleiche Länge wie der ursprüngliche text von T.
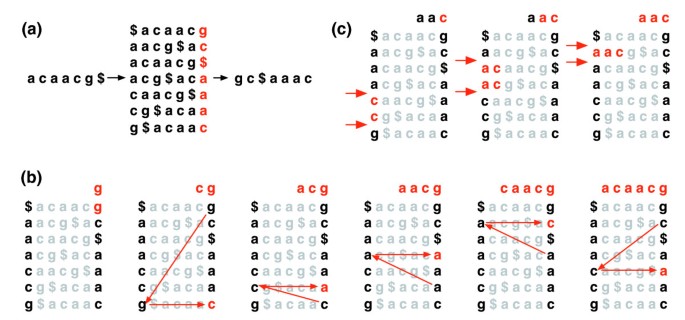
die Burrows-Wheeler-Transformation. (a) Die Burrows-Wheeler-Matrix und Transformation für „acaacg“. (b) Schritte, die EXACTMATCH unternimmt, um den Zeilenbereich und damit den Satz von Referenzsuffixen mit dem Präfix “ aac “ zu identifizieren., (c) UNPERMUTE wendet wiederholt die letzte erste (LF) Zuordnung an, um den Originaltext (in der obersten Zeile rot) aus der Burrows-Wheeler-Transformation (in der rechten Spalte schwarz) wiederherzustellen.
Diese matrix hat eine Eigenschaft namens ‚der Letzte erste (LF) mapping‘. Das ith-Vorkommen des Zeichens X in der letzten Spalte entspricht demselben Textzeichen wie das ith-Vorkommen von X in der ersten Spalte. Diese Eigenschaft unterliegt Algorithmen, die den BWT-Index zum Navigieren oder Durchsuchen des Textes verwenden., Abbildung 1b veranschaulicht UNPERMUTE, einen Algorithmus, der die LF-Zuordnung wiederholt anwendet, um T aus BWT(T) neu zu erstellen.
Das LF-Mapping wird auch in der exakten Übereinstimmung verwendet. Da die Matrix lexikographisch sortiert ist, erscheinen Zeilen, die mit einer bestimmten Sequenz beginnen, nacheinander. In einer Reihe von Schritten berechnet der EXACTMATCH-Algorithmus (Abbildung 1c) den Bereich der Matrixzeilen, der mit nacheinander längeren Suffixen der Abfrage beginnt. Bei jedem Schritt schrumpft oder bleibt die Größe des Bereichs gleich., Wenn der Algorithmus abgeschlossen ist, entsprechen Zeilen, die mit S0 (der gesamten Abfrage) beginnen, den genauen Vorkommen der Abfrage im Text. Wenn der Bereich leer ist, enthält der Text die Abfrage nicht. UNPERMUTE ist auf Burrows und Wheeler und EXACTMATCH zu Ferragina und Manzini zurückzuführen . Siehe Zusätzliche Datendatei 1 (ergänzende Diskussion 1) für Details.
Suche nach ungenauen Ausrichtungen
EXACTMATCH reicht für eine kurze Leseausrichtung nicht aus, da Ausrichtungen Fehlanpassungen enthalten können, die auf Sequenzierungsfehler, echte Unterschiede zwischen Referenz-und Abfrageorganen oder beides zurückzuführen sein können., Wir führen einen Ausrichtungsalgorithmus ein, der eine Backtracking-Suche durchführt, um schnell Ausrichtungen zu finden, die einer bestimmten Ausrichtungsrichtlinie entsprechen. Jedes Zeichen in einem Lesevorgang hat einen numerischen Qualitätswert, wobei niedrigere Werte eine höhere Wahrscheinlichkeit eines Sequenzierungsfehlers anzeigen. Unsere Ausrichtungspolitik erlaubt eine begrenzte Anzahl von Nicht Übereinstimmungen und bevorzugt Ausrichtungen, bei denen die Summe der Qualitätswerte an allen nicht übereinstimmenden Positionen niedrig ist.
Die Suche verläuft ähnlich wie EXACTMATCH und berechnet Matrixbereiche für nacheinander längere Abfragesuffixe., Wenn der Bereich leer wird (ein Suffix tritt im Text nicht auf), kann der Algorithmus eine bereits übereinstimmende Abfrageposition auswählen und dort eine andere Basis ersetzen, wodurch eine Nichtübereinstimmung in die Ausrichtung eingeführt wird. Die EXACTMATCH-Suche wird kurz nach der ersetzten Position fortgesetzt. Der Algorithmus wählt nur die Substitutionen aus, die mit der Ausrichtungsrichtlinie übereinstimmen und ein modifiziertes Suffix ergeben, das mindestens einmal im Text vorkommt. Wenn es mehrere Kandidatensubstitutionspositionen gibt, wählt der Algorithmus gierig eine Position mit einem minimalen Qualitätswert aus.,
Backtracking-Szenarien spielen sich im Kontext einer Stapelstruktur ab, die wächst, wenn eine neue Substitution eingeführt wird, und schrumpft, wenn der Aligner alle Kandidatenausrichtungen für die derzeit auf dem Stapel befindlichen Substitutionen ablehnt. Siehe Abbildung 2 für eine Illustration, wie die Suche fortgesetzt werden könnte.
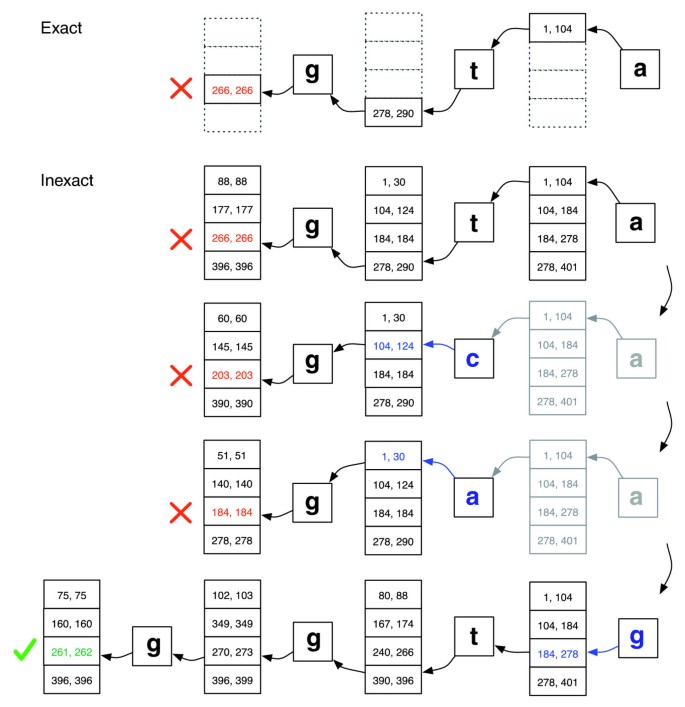
Exakte Übereinstimmung versus ungenaue Ausrichtung., Illustration, wie EXACTMATCH (top) und Bowtie ’s aligner (bottom) vorgehen, wenn es keine genaue Übereinstimmung für die Abfrage‘ ggta ‚gibt, aber es gibt eine One-Mismatch-Ausrichtung, wenn‘ a ‚durch’g‘ ersetzt wird. Boxed Zahlenpaare bezeichnen Bereiche von Matrixzeilen, die mit dem bis zu diesem Punkt beobachteten Suffix beginnen. Ein rotes X markiert, wo der Algorithmus auf einen leeren Bereich trifft und entweder abbricht (wie in EXACTMATCH) oder zurückverfolgt (wie im inexact Algorithmus). Ein grünes Häkchen, bei dem der Algorithmus einen nicht übereinstimmenden Bereich findet, der ein oder mehrere Vorkommen einer meldepflichtigen Ausrichtung für die Abfrage begrenzt.,
Kurz gesagt, Bowtie führt eine qualitätsbewusste, gierige, randomisierte Tiefensuche durch den Raum möglicher Ausrichtungen. Wenn eine gültige Ausrichtung vorhanden ist, wird Bowtie sie finden (vorbehaltlich der im folgenden Abschnitt beschriebenen Backtrack-Obergrenze). Da die Suche gierig ist, ist die erste gültige Ausrichtung, auf die Bowtie stößt, nicht unbedingt die „beste“ in Bezug auf die Anzahl der Nichtübereinstimmungen oder die Qualität., Der Benutzer kann Bowtie anweisen, die Suche fortzusetzen, bis nachgewiesen werden kann, dass jede von ihm gemeldete Ausrichtung in Bezug auf die Anzahl der Nichtübereinstimmungen „am besten“ ist (mit der Option –best). Nach unserer Erfahrung ist dieser Modus zwei-bis dreimal langsamer als der Standardmodus. Wir erwarten, dass der schnellere Standardmodus für große Re-Sequenzierungsprojekte bevorzugt wird.
Der Benutzer kann sich auch für Bowtie entscheiden, um alle Ausrichtungen bis zu einer bestimmten Anzahl (Option-k) oder alle Ausrichtungen ohne Begrenzung der Anzahl (Option-a) für einen bestimmten Lesevorgang zu melden., Wenn Bowtie im Verlauf seiner Suche N mögliche Ausrichtungen für einen bestimmten Satz von Ersetzungen findet, der Benutzer jedoch nur K Ausrichtungen angefordert hat, bei denen K < N, meldet Bowtie K der zufällig ausgewählten N Ausrichtungen. Beachten Sie, dass diese Modi viel langsamer als die Standardeinstellung sein können. Nach unserer Erfahrung ist zum Beispiel-k 1 mehr als doppelt so schnell wie-k 2.
Exzessives Backtracking
Der bisher beschriebene Aligner kann in einigen Fällen auf Sequenzen stoßen, die exzessives Backtracking verursachen., Dies tritt auf, wenn der Aligner den größten Teil seines mühelosen Backtrackings an Positionen nahe dem 3′ – Ende der Abfrage ausgibt. Bowtie mildert übermäßiges Backtracking mit der neuartigen Technik der „doppelten Indexierung“. Es werden zwei Indizes des Genoms erstellt: einer enthält den BWT des Genoms, den sogenannten „Vorwärts“ – Index, und ein zweiter enthält den BWT des Genoms mit seiner umgekehrten (nicht umgekehrten) Zeichensequenz, die als „Spiegel“ – Index bezeichnet wird. Um zu sehen, wie dies hilft, sollten Sie eine Übereinstimmungsrichtlinie berücksichtigen, die eine Nichtübereinstimmung in der Ausrichtung zulässt., Eine gültige Ausrichtung mit einer Nichtübereinstimmung fällt in einen von zwei Fällen, nach denen die Hälfte des Lesens die Nichtübereinstimmung enthält. Bowtie verläuft in zwei Phasen, die diesen beiden Fällen entsprechen. Phase 1 lädt den Vorwärtsindex in den Speicher und ruft den Aligner mit der Einschränkung auf, die er möglicherweise nicht an Positionen in der rechten Hälfte der Abfrage ersetzt. Phase 2 verwendet den Spiegelindex und ruft den Aligner für die umgekehrte Abfrage auf, mit der Einschränkung, dass der Aligner möglicherweise nicht an Positionen in der rechten Hälfte der umgekehrten Abfrage (der linken Hälfte der ursprünglichen Abfrage) ersetzt., Die Einschränkungen beim Backtracking in die rechte Hälfte verhindern ein übermäßiges Backtracking, während die Verwendung von zwei Phasen und zwei Indizes die volle Empfindlichkeit beibehält.
Leider ist es nicht möglich, ein übermäßiges Backtracking vollständig zu vermeiden, wenn Ausrichtungen zwei oder mehr Nichtübereinstimmungen aufweisen dürfen. In unseren Experimenten haben wir beobachtet, dass ein übermäßiges Backtracking nur dann signifikant ist, wenn ein Lesevorgang viele Positionen von geringer Qualität aufweist und sich nicht schlecht an der Referenz ausrichtet oder ausrichtet., Diese Fälle können mehr als 200 Backtracks pro Lesevorgang auslösen, da viele legale Kombinationen von Positionen von geringer Qualität untersucht werden müssen, bevor alle Möglichkeiten ausgeschöpft sind. Wir mindern diese Kosten, indem wir die Anzahl der Backtracks begrenzen, die zulässig sind, bevor eine Suche beendet wird (Standard: 125). Der Grenzwert verhindert, dass einige legitime, minderwertige Ausrichtungen gemeldet werden, aber wir erwarten, dass dies ein wünschenswerter Kompromiss für die meisten Anwendungen ist.,
Phased Maq-like search
Mit Bowtie kann der Benutzer die Anzahl der zulässigen Fehlanpassungen (Standard: zwei) am hochwertigen Ende eines Lesevorgangs (Standard: die ersten 28 Basen) sowie den maximal akzeptablen Qualitätsabstand der Gesamtausrichtung (Standard: 70) auswählen. Es wird angenommen , dass Qualitätswerte der Definition in PHRED folgen, wobei p die Fehlerwahrscheinlichkeit und Q = -10 ist. p.
Sowohl das gelesene als auch sein umgekehrtes Komplement sind Kandidaten für die Ausrichtung an der Referenz. Zur Verdeutlichung berücksichtigt diese Diskussion nur die Vorwärtsorientierung., Siehe zusätzliche Datendatei 1 (ergänzende Diskussion 2) für eine Erklärung, wie das umgekehrte Komplement integriert wird.
Die ersten 28 Basen am hochwertigen Ende des Lesevorgangs werden als „Samen“ bezeichnet. Das Saatgut besteht aus zwei Hälften: dem 14 bp am hochwertigen Ende (normalerweise dem 5′-Ende) und dem 14 bp am minderwertigen Ende, das als „Hi-half“ bzw., Unter der Annahme der Standardrichtlinie (zwei im Seed zulässige Nichtübereinstimmungen) fällt eine meldepflichtige Ausrichtung in einen von vier Fällen: keine Nichtübereinstimmungen in Seed (Fall 1); keine Nichtübereinstimmungen in Hi-half, ein oder zwei Nichtübereinstimmungen in lo-half (Fall 2); keine Nichtübereinstimmungen in lo-half, ein oder zwei Nichtübereinstimmungen in Hi-Half (Fall 3); und eine Nichtübereinstimmung in Hi-half, eine Nichtübereinstimmung in lo-half (Fall 4).
Alle Fälle erlauben eine beliebige Anzahl von Fehlanpassungen im Nonseed-Teil des Lesens und alle Fälle unterliegen ebenfalls der Qualitätsabstandsbeschränkung.,
Der Bowtie-Algorithmus besteht aus drei Phasen, die zwischen der Verwendung des Vorwärts-und Spiegelindizes wechseln, wie in Abbildung 3 dargestellt. Phase 1 verwendet den Spiegelindex und ruft den Aligner auf, um Ausrichtungen für die Fälle 1 und 2 zu finden. Phasen 2 und 3 arbeiten zusammen, um Ausrichtungen für Fall 3 zu finden: Phase 2 findet Teilausrichtungen mit Fehlanpassungen nur in der Hi-Hälfte und Phase 3 versucht, diese Teilausrichtungen auf Vollausrichtungen auszudehnen. Schließlich ruft Phase 3 den Aligner auf, um Ausrichtungen für Fall 4 zu finden.,

Die drei Phasen des Bowtie-Algorithmus für die Maq-Politik. Ein dreiphasiger Ansatz findet Ausrichtungen für Zwei-Mismatch-Fälle 1 bis 4 und minimiert gleichzeitig das Backtracking. Phase 1 verwendet den Spiegelindex und ruft den Aligner auf, um Ausrichtungen für die Fälle 1 und 2 zu finden. Phasen 2 und 3 arbeiten zusammen, um Ausrichtungen für Fall 3 zu finden: Phase 2 findet nur in der Hi-Hälfte Teilausrichtungen mit Fehlanpassungen, und Phase 3 versucht, diese Teilausrichtungen auf Vollausrichtungen auszudehnen., Schließlich ruft Phase 3 den Aligner auf, um Ausrichtungen für Fall 4 zu finden.
Performance-Ergebnisse
Wir bewerteten die Leistung von Bowtie mit liest aus dem 1.000-Genome-Projekt pilot (Nationales Zentrum für Biotechnologie-Informationen Kurz Archiv Lesen:SRR001115). Insgesamt 8,84 Millionen Lesevorgänge, etwa eine Spur von Daten eines Illumina-Instruments, wurden auf 35 bp getrimmt und auf das menschliche Referenzgenom ausgerichtet . Sofern nicht anders angegeben, werden die gelesenen Daten nicht gefiltert oder geändert (außer Trimmen), wie sie im Archiv erscheinen., Dies führt zu etwa 70% bis 75% der Lesevorgänge, die sich irgendwo am Genom ausrichten. Nach unserer Erfahrung ist dies typisch für Rohdaten aus dem Archiv. Aggressivere Filterung führt zu höheren Ausrichtungsraten und schnellerer Ausrichtung.
Alle Läufe wurden auf einer einzigen CPU ausgeführt. Bowtie Speedups wurden als Verhältnis der Wanduhr-Ausrichtungszeiten berechnet. Sowohl die Wanduhr als auch die CPU-Zeiten werden angegeben, um zu demonstrieren, dass die Eingabe – /Ausgabelastung und der CPU-Streit keine signifikanten Faktoren sind.
Die Zeit, die zum Erstellen des Bowtie-Index benötigt wurde, war nicht in den Bowtie-Laufzeiten enthalten., Im Gegensatz zu konkurrierenden Tools kann Bowtie einen vorberechneten Index für das Referenzgenom in vielen Ausrichtungsläufen wiederverwenden. Wir gehen davon aus, dass die meisten Benutzer solche Indizes einfach aus einem öffentlichen Repository herunterladen werden. Die Bowtie-Site bietet Indizes für aktuelle Builds der Genome Mensch, Schimpanse, Maus, Hund, Ratte und Arabidopsis thaliana sowie vieler anderer.
Die Ergebnisse wurden auf zwei Hardwareplattformen erzielt: einer Desktop-Workstation mit 2,4 GHz Intel Core 2-Prozessor und 2 GB RAM; und ein Großspeicherserver mit einem 2,4 GHz AMD Opteron-Prozessor mit vier Kernen und 32 GB RAM., Diese werden mit “ PC „bzw.“ Server “ bezeichnet. Sowohl PC als auch Server führen Red Hat Enterprise Linux ALS Release 4 aus.
Vergleich zu SOAP und Maq
Maq ist ein beliebter Aligner, der zu den am schnellsten konkurrierenden Open-Source-Tools zum Ausrichten von Millionen von Illumina-Lesevorgängen am menschlichen Genom gehört. SOAP ist ein weiteres Open-Source-Tool, das in kurzgelesenen Projekten gemeldet und verwendet wurde .
Tabelle 1 zeigt die Leistung und Empfindlichkeit von Bowtie v0.9.6, SOAP v1.10 und Maq v0.6.6. SOAP konnte nicht auf dem PC ausgeführt werden, da der Speicherbedarf von SOAP den physischen Speicher des PCS übersteigt. Die ‚ Seife.,contig ‚ Version der SOAP-Binärdatei wurde verwendet. Zum Vergleich mit SOAP wurde Bowtie mit ‚-v 2 ‚ aufgerufen, um die standardmäßige Übereinstimmungsrichtlinie von SOAP nachzuahmen (die bis zu zwei Nichtübereinstimmungen in der Ausrichtung zulässt und Qualitätswerte ignoriert), und mit ‚–maxns 5‘, um die Standardrichtlinie von SOAP zum Herausfiltern von Lesevorgängen mit fünf oder mehr Vertrauensbasen ohne Vertrauen zu simulieren. Für den Maq-Vergleich wird Bowtie mit seiner Standardrichtlinie ausgeführt, die die Standardrichtlinie von Maq nachahmt, bis zu zwei Nichtübereinstimmungen in den ersten 28 Basen zuzulassen und ein Gesamtlimit von 70 für die Summe der Qualitätswerte an allen nicht übereinstimmenden Lesepositionen durchzusetzen., Um den Speicherbedarf von Bowtie besser mit dem von Maq vergleichbar zu machen, wird Bowtie in allen Experimenten mit der Option ‚-z‘ aufgerufen, um sicherzustellen, dass nur der Vorwärts-oder Spiegelindex gleichzeitig im Speicher verbleibt.
Die Anzahl der ausgerichteten Lesevorgänge zeigt an, dass SOAP (67.3%) und Bowtie-v 2 (67.4%) eine vergleichbare Empfindlichkeit aufweisen. Von den Lesungen, die entweder von SOAP oder Bowtie ausgerichtet wurden, wurden 99,7% von beiden ausgerichtet, 0,2% wurden von Bowtie, aber nicht von SOAP, und 0,1% wurden von SOAP, aber nicht Bowtie ausgerichtet. Maq (74.7%) und Bowtie (71.9%) haben ebenfalls eine ungefähr vergleichbare Empfindlichkeit, obwohl Bowtie um 2.8% zurückbleibt., Von den Lesungen, die entweder von Maq oder Bowtie ausgerichtet wurden, wurden 96.0% von beiden ausgerichtet, 0.1% wurden von Bowtie, aber nicht Maq, und 3.9% wurden von Maq, aber nicht Bowtie ausgerichtet. Von den Lesevorgängen, die von Maq, aber nicht von Bowtie abgebildet werden, sind fast alle auf eine Flexibilität im Ausrichtungsalgorithmus von Maq zurückzuführen, die es einigen Ausrichtungen ermöglicht, drei Fehlanpassungen im Seed zu haben. Der Rest der Lesevorgänge, die von Maq, aber nicht von Bowtie, abgebildet werden,ist auf Bowties Rückverfolgbarkeit zurückzuführen.
In der Dokumentation von Maq wird erwähnt, dass Lesevorgänge, die „Poly-A-Artefakte“ enthalten, die Leistung von Maq beeinträchtigen können., Tabelle 2 zeigt die Leistung und Empfindlichkeit von Bowtie und Maq, wenn der Lesesatz mit dem Befehl ‚catfilter‘ von Maq gefiltert wird, um Poly-A-Artefakte zu eliminieren. Der Filter eliminiert 438.145 von 8.839.010 Lesevorgängen. Andere experimentelle Parameter sind identisch mit denen der Experimente in Tabelle 1, und die gleichen Beobachtungen über die relative Empfindlichkeit von Bowtie und Maq gelten hier.
Leselänge und Leistung
Mit der Verbesserung der Sequenzierungstechnologie wachsen die Leselängen über die heute in öffentlichen Datenbanken üblichen 30 bis 50 bp hinaus., Bowtie, Maq und SOAP unterstützen Lesevorgänge mit Längen von bis zu 1.024, 63 und 60 bp, und die Maq-Versionen 0.7.0 und höher unterstützen Leselängen von bis zu 127 bp. Tabelle 3 zeigt die Leistungsergebnisse, wenn die drei Werkzeuge jeweils verwendet werden, um drei Sätze von 2 M ungestrimmten Lesevorgängen, einen 36-BP-Satz, einen 50-BP-Satz und einen 76-BP-Satz, am menschlichen Genom auf der Serverplattform auszurichten. Jeder Satz von 2 M wird zufällig aus einem größeren Satz entnommen (NCBI Short Read Archive: SRR003084 für 36-bp, SRR003092 für 50-bp, SRR003196 für 76-bp)., Lesevorgänge wurden so abgetastet, dass die drei Sätze von 2 M eine einheitliche Fehlerrate pro Basis aufweisen, wie sie aus Phred-Qualitäten pro Basis berechnet wird. Alle Lesevorgänge gehen durch Maq ‚ s ‚catfilter‘.
Bowtie wird sowohl im Maq-ähnlichen Standardmodus als auch im SOAP-ähnlichen ‚-v 2‘ – Modus ausgeführt. Bowtie erhält auch die Option ‚- z‘, um sicherzustellen, dass nur der Forward-oder Mirror-Index gleichzeitig im Speicher gespeichert ist. Maq v0.7.1 wurde anstelle von Maq v0.6.6 für den 76-bp-Satz verwendet, weil v0.6.,6 liest nicht länger als 63 bp ausrichten. SOAP wurde nicht auf dem 76-BP-Set ausgeführt, da Lesevorgänge länger als 60 bp nicht unterstützt werden.
Die Ergebnisse zeigen, dass der Maq-Algorithmus insgesamt auf längere Leselängen besser skaliert als Bowtie oder SOAP. Bowtie im SOAP-like ‚-v 2‘ – Modus skaliert jedoch auch sehr gut. Bowtie skaliert in seinem standardmäßigen Maq-ähnlichen Modus gut von 36-bp auf 50-bp-Lesevorgänge, ist jedoch bei 76-BP-Lesevorgängen wesentlich langsamer, obwohl es immer noch mehr als eine Größenordnung schneller als Maq ist.,
Parallele Leistung
Die Ausrichtung kann parallelisiert werden, indem Lesevorgänge auf gleichzeitige Suchthreads verteilt werden. Mit Bowtie kann der Benutzer eine gewünschte Anzahl von Threads angeben (Option-p); Bowtie startet dann die angegebene Anzahl von Threads mithilfe der pthreads-Bibliothek. Bowtie-Threads synchronisieren sich beim Abrufen von Lesevorgängen, beim Ausgeben von Ergebnissen, beim Umschalten zwischen Indizes und beim Ausführen verschiedener Formen der globalen Buchhaltung, z. B. beim Markieren eines Lesevorganges als „fertig“., Andernfalls können Threads parallel betrieben werden, was die Ausrichtung auf Computern mit mehreren Prozessorkernen erheblich beschleunigt. Das Speicherabbild des Index wird von allen Threads gemeinsam genutzt, sodass der Platzbedarf nicht wesentlich ansteigt, wenn mehrere Threads verwendet werden. Tabelle 4 zeigt die Leistungsergebnisse für die Ausführung von Bowtie v0.9.6 auf dem Vier-Kern-Server mit einem, zwei und vier Threads.,
Index Gebäude
Bowtie verwendet eine flexible indexing-Algorithmus, die können konfiguriert werden, um den trade-off zwischen Speichernutzung und Laufzeit. Tabelle 5 veranschaulicht diesen Kompromiss bei der Indizierung des gesamten menschlichen Referenzgenoms (NCBI Build 36.3, contigs). Läufe wurden auf der Serverplattform durchgeführt. Der Indexer wurde viermal mit unterschiedlichen Obergrenzen für die Speichernutzung ausgeführt.,
Die gemeldeten Zeiten vergleichen günstig mit Ausrichtung Zeiten des konkurrierenden tools, führen Sie die Indizierung während der Ausrichtung. Weniger als 5 Stunden sind für Bowtie erforderlich, um einen Whole-Human-Index mit 8.84 Millionen Lesevorgängen aus dem 1,000 Genome Project (NCBI Short Read Archive:SRR001115) auf einem Server zu erstellen und abzufragen, mehr als sechsmal schneller als der entsprechende Maq-Lauf., Die unterste Zeile zeigt, dass der Bowtie Indexer mit entsprechenden Argumenten speichereffizient genug ist, um auf einer typischen Workstation mit 2 GB RAM ausgeführt zu werden. Zusätzliche Datendatei 1 (Ergänzende Diskussionen 3 und 4) erläutert den Algorithmus und den Inhalt des resultierenden Index.
Software
die Fliege ist in C++ geschrieben und verwendet die SeqAn-Bibliothek . Der Konverter in das Maq-Mapping-Format verwendet Code von Maq.