Indledning
Substituent effekter udgør et centralt begreb for forståelse af reaktivitet og spektroskopiske adfærd organiske forbindelser (Krygowski & St??pie,, 2005). I en simpel tilgang kan substituentvirkninger klassificeres i henhold til mekanismen for interaktion med det reaktive center som induktive (gennem σ-bindinger) eller resonanseffekter (gennem π-bindinger)., Ikke desto mindre vil nogle yderligere udtryk (såsom steriske, felt-eller opløsningsmiddelvirkninger) være nødvendige for en grundig beskrivelse af substituentvirkninger.
Da Ingold ‘s klassificering af elektroniske substituent effekter (Ingold, 1953), den alkyl-gruppen har været betragtet som en σ-donor substituent (+jeg, i Ingold’ s nomenklatur) i de fleste Organiske Kemi lærebøger (Burrows, Holman, Parsons, Pilling, & Pris, 2013; Hornback, 2006; Roos & Roos, 2014; Smith, 2013; Vollhardt & Schore, 2014)., Ikke desto mindre bør EEE ‘ s kritik af et så forenklet synspunkt bemærkes:
“i vand er propansyre lidt svagere end eddikesyre. Arten af den induktive virkning af en alkylgruppe diskuteres af kemikere. Alkylgrupper stabilisere carbocations og i denne rolle synes at være elektron-frigivende. De øger også basiteten af aminer, hvilket igen tyder på, at de er elektronfrigørende. På den anden side, selvom tert-butylalkohol (PKA 19) er en svagere syre end ethanol (PKA 17) i vand, er den stærkere syre i gasfasen., Denne eksperimentelle observation antyder, at alkylgrupper kan stabilisere anioner såvel som kationer, og at solvation spiller en vigtig rolle i bestemmelsen af relative syrer. Derfor er en advarsel nødvendig. De relative syrheder, som generaliseringerne præsenteret i dette kapitel er baseret på, blev bestemt i vand. I gasfasen ses ofte reverseringer i rækkefølge af beslægtede forbindelser.”(EEE, 1999, s. 107)
nogle alkylsubstitutionseffekter er ofte blevet forklaret i lærebøger på modstridende eller gådefulde måder., Således, kemisk skift forskelle mellem og CH2 CH3-grupper er tilskrevet i Hornback ‘ s book til det faktum, at “carbon er lidt mere elektronegative end brint” (Hornback, 2006, s. 549) på trods af alkyl-gruppe, der tidligere har været klassificeret som en svag induktiv elektron-donere substituent (Hornback, 2006, s. 117). I Vollhardt ‘ s lærebog, forholdet mellem methyl-gruppe kemiske skift for en række CH3X forbindelser og X elektronegativiteten er illustreret i en tabel, der mangler en løsning til X=methyl (Vollhardt & Schore, 2014, s., 389), hvorved det ubekvemme kulstofproblem undgås.
Jeg viser her, at alkylgruppen opfører sig som A –i+R-substituent. Selvom nogle faktorer (såsom felt -, steriske eller opløsningsmiddelvirkninger) implicit ignoreres i denne tilgang, kan mange aktuelt tilgængelige teoretiske og eksperimentelle beviser således beskrives på en nem måde.
En Cδ–Hδ+ bond polarisering er blevet eksperimentelt observeret for metan (Lazzeretti, Zanasi, & Raynes, 1987), i overensstemmelse med den større elektronegativiteten af kulstof i forhold til brint, 2.55 vs. 2.,20 i Pauling-skalaen (Allred, 1961). Et sådant polarisationsmønster gør det muligt at forudsige dipolmomentretningen for enkle kulbrinter gennem additive modeller, selvom kvantitativ aftale normalt er beskeden (2-methylpropan: 0.3 D estimeret vs. 0.132 D eksperimentel) (Dean, 1999).
da hydrogen anvendes som standard i Ingolds klassificering af substituenter (Krygo ?ski & St??pie., 2005), bør alkylgruppen klassificeres som A –i-substituent (deraf en electr-elektronudtagningsgruppe). En sådan rolle er illustreret i Fig., 1 For bond-bindingspolarisering fra uanset atom Y til en methylgruppe, selvom omvendt bindingspolarisering forventes, når Y er et mere elektronegativt end kulstof (f.chlor).
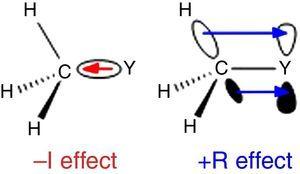
– i (venstre) og +R(højre) virkninger af en methylgruppe bundet til et atom Y.
En anden adfærd er fundet til alkyl-grupper, når der er knyttet til sp2 eller sp-hybridiseret atomer på grund af elektron densitet donation fra alkyl-C–H eller C–C σ obligationer til tom p orbital af den tilstødende atom (den enkleste π-system), som vist i Fig. 1. Således faldet af gas-fase surhedsgrad for phenol og benzoesyre gennem s-methyl substitution (McMahon & Kebarle, 1977) kan kun henføres til en betydelig π-donor virkning for methyl substituent (ja, større end for methoxy-gruppen)., Alkylgruppen bør imidlertid betragtes som en atypisk π-donorsubstituent på grund af manglen på ensomme elektronpar. Sådan en σ-bond/π-system interaktion, som er opkaldt som hyperconjugation (Mullins, 2012) kan umiddelbart forklares ved en analogi med π-donor adfærd af en enlig par-bærende atom (fx, chlor) til en tom p orbital, selvom C–C eller C–H-obligationer (snarere end electron lone par) af alkyl-gruppen er involveret som elektron-releasing enheder i hyperconjugative interaktioner., Interessant nok er interactions* * * interaktioner (negativ hyperconjugation) normalt ubetydelige for alkylgrupper, der mangler elektronegative atomer (Bocca, Pontes, & Basso, 2004).
nogle molekylære strukturelle træk kan rationaliseres på basis af alkylgruppeegenskaberne. For eksempel er de større CO-bindingslængder, der findes i methylketoner (acetone: e .p. 1.210 Å, calc. 1.193 Å) i sammenligning med de beslægtede aldehyder (acetaldehyd: e .p. 1.209 Å, calc. 1.,188Å) (Berry, Toiletartikler, Pacansky, & Hagler, 1995), der kan henføres til stabilisering af zwitterionic resonans form (se Fig. 2) gennem alkylgruppe π-donation til det carbonylliske carbonatom, hvilket svækker carbonylgruppens dobbeltbindingsfunktion.
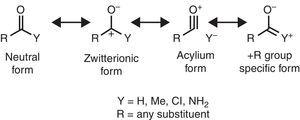
neutrale (venstre) og rightititterioniske (højre) resonansformer af en carbonylforbindelse.
Hyperconjugative interaktioner er afhængig af placeringen af C–H (eller C–C) obligationer i forhold til p-orbital af den tilstødende atom Y, den mest effektive samspil, der svarer til en næsten parallel arrangement. For eksempel, toluen Csp3–H bond næsten vinkelret på den ramme flyet er lidt længere end de andre Csp3–H-obligationer (af 0.002 Å, Hameka & Jensen, 1996)., Geometri afhængighed af hyperconjugation giver mulighed for at forklare konformationelle analyse af methyl-substituerede umættede forbindelser, såsom propen (Liberles, O ‘ Leary, Eilers, & Whitman, 1972) eller acetaldehyd (Muñoz-Caro, Niño, & Moule, 1994).
Som en velkendt konsekvens af π-donor opførsel af alkyl-gruppen, alkyl substitution giver mere elektron-rige alkener og arenes (Libit & Hoffmann, 1974)., Den høje reaktivitet af en alkylsubstitueret aren i en Searreaktion kan således tilskrives stabiliseringen af det tilsvarende Hvalland-mellemprodukt gennem π-elektrondonation.alkylgruppens-i+R-opførsel gør det muligt at forklare en række træk ved alkylsubstituerede forbindelser, såsom dipolmomenter, spektroskopiske egenskaber og reaktivitet (i gasfase og opløsningsmedier), som vist nedenfor.
dipolmomenter
alkylgruppens elektron-tilbagetrækningsadfærd i alifatiske forbindelser afspejles også i dipolmomenter., Således er dipolmomentvektorerne for propan og 2-methylpropan (Tasi et al., 1997), såvel som nogle substituerede cyclooctaner (B .hm & e .ner, 2004) kan tilskrives den tilbagetrækningseffekt (–I) af methylgruppen i sammenligning med hydrogen (se Fig. 3).
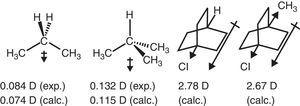
dipolmomenter af propan og substituerede cyclooctaner.
I modsætning, det π-donor karakter af methyl-gruppe (+R) er påkrævet for at forklare rejse dipol øjeblikke af nitrobenzen og benzonitrile gennem s-methyl substitution (Brown, 1959) (se Fig. 4).
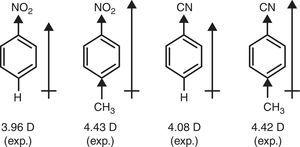
dipolmomenter af ben .enderivater.
molekylære dipolmomenter kan beregnes pålideligt ved hjælp af aktuelle beregningsmetoder., Interessant nok er de beregnede dipolmomentvektorer for et sæt enkle kulbrinter (Tasi et al., 1997) har tilladt at udlede en dobbelt rolle for methylgruppen: elektron-tilbagetrækning, når den er bundet til sp3-carbonatomer, men elektrondonerende, når den er bundet til sp2-eller sp3-carbonatomer.
en sådan dobbelt opførsel af alkylsubstituenten observeres også for heteroatombærende forbindelser. Således en gradvis dipol øjeblik fald er observeret for efterfølgende methyl substitution på ammoniak (NH3, 1.47 D; MeNH2, 1.31 D; Me2NH, 1.01 D; Me3N, 0.,61 D) (Le Fèvre & Russell, 1947), i overensstemmelse med den gradvise formindskelse af kvælstof elektron densitet (Hehre & Pople, 1970). I modsætning, en dipol øjeblik ekstraudstyr (fra 1.53 D til 1.68 D) (Nelson, Lide, & Maryott, 1967) er fundet på for N,N-dimethyl substitution på anilin (Targema, Obi-Egbedi, & Adeoye, 2013), i overensstemmelse med den hæve i π-donor karakter for amino gruppe (Hinchliffe & Kidd, 1980) på grund +R-bidrag af methyl substituenter.,spektroskopiske egenskaber spektroskopiske egenskaber af mange organiske forbindelser kan let rationaliseres ved at antage A –i+R-adfærd for alkylgruppen som et generelt træk. Således kan NMR – kemisk forskydning af et atom betragtes som et eksperimentelt mål for elektrondensiteten ved den tilsvarende kerneposition, selvom andre effekter – såsom anisotrope magnetfelter-også kan være involveret. Do .nfield skift induceret af en methylsubstituent på sp3 carbonatomer (+9,6 ppm i 13C NMR) eller de tilsvarende bundne hydrogenatomer (+0.,63ppm i 1H NMR) (Pretsch, Bühlmann, & Badertscher, 2009), er i overensstemmelse med den adfærd, typisk i grupper (såsom halogenatomer).
Alkyl substitutionseffekter på NMR-kemiske skift af alkener vis en elektron densitet fald i α position (+12.9 ppm for 13C NMR; +0.45 ppm for 1H NMR), samt en tæthed hæve i β position (-7.4 ppm for 13C; -0.31/-0.40 ppm for 1H), i overensstemmelse med en –i+R-effekt, selvom anisotropisk effekter (såsom ring strøm) kan også spille en rolle. En sådan-i+R-adfærd findes også for alkyner ifølge 13C NMR-spektroskopi (+8.,5 ppm for α position, -3.6 ppm for β position).
den dikotomiske opførsel af alkylsubstituenter på π-systemer (elektrondensitetshøjde for α-atom, elektrondensitetsfald for β-atom) kan ikke forklares på grundlag af en simpel opførsel (såsom A +I-effekt).
En –i+R-adfærd (Meier, 2007) er observeret gennem 15N-NMR-spektroskopi til alkyl substitution på aminer og amider afhængigt af kvælstof hybridisering (downfield skift til alifatiske aminer, upfield skift til Nsp2-bærende stoffer – såsom anilines og amider).,
NMR-koblingskonstanter er også afhængige af substituerende elektroniske egenskaber (såvel som nogle geometriske egenskaber). Således er der en betydelig nedgang er fundet for 1H–1H kobling konstanter gennem methyl substitution (trans, -2.3 Hz; cis, -1.6 Hz; perle, -0.4 Hz), i kvalitative aftale med data fra en typisk elektron-fratagelse af grupper, såsom fluoratom (trans, -6.3 Hz; cis, -6.9 Hz; perle, -5.7 Hz). Det positive bidrag til methylsubstitution på 13C-1H koblingskonstanter af alifatiske forbindelser (+1.,0h.), er også kvalitativt i overensstemmelse med dem fra andre –i-grupper (fluor, +24h.).
infrarød spektroskopi er også følsom over for substituentegenskaber, som illustreret ved Co-strækningsfrekvensen af carbonylforbindelser som en funktion af den tilsvarende substituent Y, som kan rationaliseres i form af resonansformer (fig. 2). Ved at tage et alifatisk aldehyd (ca., 1725cm–1) som en reference, er den rødforskydning (bølgetal fald), der er fremkaldt af a +jeg substituent (acetyltrimethylsilane, 1645cm–1: Soderquist & Hsu, 1982) kan henføres til stabilisering af zwitterionic form. I stedet openworld provokeret af en –jeg substituent (reaktion), >1800cm–1: Pretsch et al., 2009) kan forklares ved hjælp af to alternative eller samtidige mekanismer (destabilisering af den AC .itterioniske form og/eller bidrag fra en acyliumionbærende form). Endelig redshifts provokeret af + R substituenter (amider, ca., 1680cm–1: Pretsch et al., 2009) kan tilskrives bidraget fra en bestemt resonansformular. Den lette rødforskydning induceret af alkylgruppe (methylketoner, ca. 1715cm-1) viser en nettoelektron-donerende effekt (dermed en overvægt af +R –effekten over-i-egenskaber). Nettet donor effekt af carbonyl-bundet alkyl-gruppen er i overensstemmelse med den større dipol moment af acetone (2.88 D) i forhold til formaldehyd (2.33 D) (Nelson et al., 1967).
alkylgruppens indflydelse på UV–Vis spektre af mange forbindelser kan også forklares med hensyn til elektroniske effekter., Således bathochromic skift, der er fremkaldt af alkylgrupper på UV-absorption bands af α,β-umættede forbindelser (+10nm i α position, +12nm i β-position), konjugeret polyenes (+5mn) eller benzen derivater (+3.0 nm) er kvalitativt i overensstemmelse med effekter af typiske π-donor grupper (fx, klor).
gasfase syre–base reaktivitet
Relative basiciteter af alifatiske aminer i vandig opløsning er blevet tilskrevet den formodede +i-effekt af alkylgruppe (Sorrell, 2006)., Interestingly, the irregular basicity order of amines in water (Me2NH>MeNH2>Me3N>NH3, as shown by the pKa values for the corresponding conjugated acids: 10.77>10.62>9.80>9.,246) (Dean, 1999) er forurenet med opløsningsmidler effekter, som illustreret ved den systematiske basicity rækkefølgen af aminer i gasfase (Me3N>Me2NH>MeNH2>NH3) (Brauman, Riveros, & Blair, 1971)., Selv om gas-fase basicity for kan henføres til normalt antages +jeg alkyl effekt (Carter 2007), en nedsættelse af kvælstof elektron densitet gennem methyl substitution har været faktisk observeret ved hjælp af Molekylære Elektrostatisk Potentiale beregninger (Baeten, De Proft, & Geerlings, 1995), hvilket tyder på en –jeg adfærd for methyl-gruppen., Faktisk bør gasfasebasicitetsordren for alifatiske aminer tilskrives den stigende stabilisering af substituerede ammoniumioner på grund af alkylgruppen polariserbarhed (Aue, diebb, & bo .ers, 1976).
i Forhold acidities af alkoholer i vandig opløsning (H2O>MeOH>EtOH>iPrOH>tBuOH) har også været tilskrevet i nogle lærebøger til den forudsatte alkyl +I effekt (Johnson, 1999; Solomons, Fryhle, & Snyder, 2016)., Da det omvendte surhedsgrad ordre er fundet i gasfasen, i forhold acidities af alkoholer i vand, bør henføres til den lavere størrelser af opløsning enthalpies for større alkoxide anioner (Brauman & Blair, 1969).
diskussionen om alkylgruppe elektroniske egenskaber kan også anvendes på carbanioner. Således “lærebog” stabilitet for enkel carbanions (methyl>ethyl>isopropyl>tert-butyl) er blevet tilskrevet den forudsatte +jeg induktiv effekt af alkylgrupper (Burrows et al.,, 2013; Chaloner, 2015; Roos & Roos, 2014; Smith, 2013). Imidlertid findes der en uregelmæssig rækkefølge for gasfase-carbanion-stabiliteter (tBu> Me > iPr >Et) i overensstemmelse med sammenfald af to modsatte alkylvirkninger (DePuy et al., 1989): en stabiliseringsmekanisme gennem alkylpolariserbarhed (det vil sige n Hyper Hyper* hyperconjugation) og en destabiliserende tendens (konsekvent med en +R-rolle ved at antage en p-lignende opførsel for det kulstof-ensomme par).,
stabiliteten af andre reaktionsmedier kan også vurderes på basis af alkylgruppeeffekter. Således, den velkendte stabilitet for vigtige forbindelser er gan (videregående>sekundære>primære>methyl) har været til tider tilskrives en positiv induktiv effekt (Chaloner, 2015; Roos & Roos, 2014)., Det er interessant, hyperconjugation er præsenteret i mange lærebøger som en alternativ forklaring til den stabilitet, orden af vigtige forbindelser er gan (Brown, Iverson, Anslyn, & Foote, 2013; Burrows et al., 2013) selvom den sædvanlige tvetydige skrivning forhindrer at fastslå, om begge forklaringer svarer til enten to forskellige beskrivelser af det samme fænomen eller to samtidige mekanismer, der spiller i samme retning., Alligevel bør stabilitetsordren for karbokationer tilskrives hyperconjugation (dermed A+R-adfærd på en ledig p-orbital, det enkleste system-system), selvom andre interaktioner (såsom alkylpolariserbarhed) også er involveret (Aue, 2011).
frie radikaler viser den samme stabilitetsordre som carbocations, hvilket indikerer stabilisering gennem alkylsubstitution. Selvom en sådan stabilitetsordre kan være berettiget på grundlag af en antaget +i-adfærd, kan +R-effekten alternativt betragtes, analogt med stabiliseringen af frie radikaler ved ensomme parbærende atomer (2006ipse, 2006).,
Reaktivitet i opløsning
i Forhold acidities af simple carboxylsyrer i vandig opløsning (eddikesyre>propionsyre>smørsyre) har været anvendt i nogle lærebøger for at illustrere den forudsatte +I kraft af alkyl-gruppen (Sorrell, 2006). Interessant, den omvendte rækkefølge er fundet, når enthalpies er i stedet betragtes (Christensen, Izatt, & Hansen, 1967), hvilket tyder på, at surhedsgraden for i vandig opløsning skal tilskrives hydrering entropies., Således signifikant gitter orden af flydende vand (fordampning entropi svarende 118,89 Jmol–1K–1, i modsætning til typiske værdier af ca. 88Jmol-1K-1 For de fleste væsker, Dean, 1999) kan indføre betydelige ændringer på reaktionsenergi. Især fører hydrering af apolære molekyler (eller dele) til en yderligere opløsningsmiddelgitterbestilling (blok .ijl & Engberts, 1993). Som en konsekvens maskeres alkylgruppeinduktive virkninger fra eksperimentelle data i vandig opløsning ofte af hydratiseringsentropier (Calder & Barton, 1971)., I forhold acidities af simple carboxylsyrer i gasfase (Yamdagni & Kebarle, 1973) og acetonitril (Eckert et al., 2009) er i overensstemmelse med den store rolle, som hydratiseringsentropier spiller.
den lavere surhed af pivalsyre i sammenligning med eddikesyre, som normalt tilskrives den antagede +i-virkning af alkylgruppen (Smith, 2008), vendes, når reaktionsenthalpies overvejes (Eckert et al., 2009).,
den formodede +i-alkylgruppeeffekt på surhedsgraden af simple Carbo .ylsyrer i vandig opløsning kan således tilskrives en artefakt afledt af opløsningsmiddelvirkninger. Mens en volumenforøgelse af neutrale opløste stoffer fører til en hydratiserings entropiforøgelse, findes det omvendte forhold for ioniske arter (Gra .iano, 2009). Som en konsekvens heraf, alkyl substitution (gennem en forøgelse af den molekylære volume), som fører til stabilisering (i Gibbs fri energi) af ikke-ioniseret syre i vand samt destabilisering af den tilsvarende carboxylat anion, hvilket medfører en surhedsgrad falder.,
Det større syreindhold af myresyre i sammenligning med eddikesyre vandig opløsning (pKa-værdier: 3.751 og 4.756, henholdsvis, Dean, 1999) har også været diskuteret i mange lærebøger som et eksempel på anvendelse af induktive effekter (Hart, Hadad, Craine, & Hart, 2012; Hornback, 2006; Okuyama & Maskill, 2014; Roos & Roos, 2014). Da meget lignende reaktion enthalpies er involveret i dissociationsreaktionerne af myresyre og eddikesyre (Christensen et al.,, 1967), skal den større surhed af myresyre faktisk tilskrives hydratiserings entropi forskelle.
konklusioner
en klar forståelse af induktive og resonanseffekter er en vigtig nøgle til en god indlæring af organisk kemi (Mullins, 2008). Overraskende nok er den næsten allestedsnærværende alkylgruppe forkert præsenteret i mange lærebøger som en donor-donor (+i) gruppe. Imidlertid vises en dobbelt opførsel af alkylsubstituenter afhængigt af hybridiseringen af naboatomet., Således, alkyl-grupper bundet til alifatiske kæder opføre sig, som σ-acceptorer (–I overensstemmelse med den større elektronegativiteten af kulstof i forhold til brint), der henviser til, at dem, der er knyttet til π-systemer fungerer som π-donorer (+R, på grund af hyperconjugative interaktioner). En række eksperimentelle og teoretiske data (dipolmomenter, NMR, IR og UV-spektre, reaktivitet) er enige i en sådan dobbelt opførsel.,
hele analysen af alle data, der overvejes her, tillader at udlede en lille –i-effekt såvel som en betydelig +R-opførsel for alkylgruppen som en funktion, der er gyldig i alle diskussioner om organiske forbindelsers spektroskopiske og reaktivitetsegenskaber.
interessekonflikt
forfatteren erklærer ingen interessekonflikt.