Úvod
Substituent účinky představují klíčový pojem pro pochopení reaktivity a spektroskopické chování organických sloučenin (Krygowski & St??pień, 2005). V jednoduchém přístupu lze substituční účinky klasifikovat podle mechanismu interakce s reaktivním centrem jako induktivní (prostřednictvím σ-vazeb) nebo rezonanční účinky (prostřednictvím π-vazeb)., Pro důkladný popis substituentních účinků by však byly vyžadovány některé další pojmy (například sterické, polní nebo solventní účinky).
Od Ingold, klasifikace elektronických substituent účinky (Ingold, 1953), alkyl skupina byla považována za σ-donor substituent (+já, v Ingold je nomenklatury) ve většině Organické Chemie učebnice (Burrows, Holman, Parsons, Žmolkování, & Cena, 2013; Hornback, 2006; Roos & Roos, 2014; Smith, 2013; Vollhardt & Schore, 2014)., Nicméně, Eğe kritiku k takové zjednodušující hledisko by mělo být poznamenal:
„Ve vodě, kyseliny propanové je mírně slabší než kyselina octová. O povaze induktivního účinku alkylové skupiny diskutují chemici. Alkylové skupiny stabilizují karbokace a v této roli se jeví jako uvolňování elektronů. Také zvyšují bazicitu aminů, což opět naznačuje, že se uvolňují elektrony. Na druhou stranu, ačkoli terc-butylalkohol (pKa 19) je slabší kyselina než ethanol (pKa 17) ve vodě, je silnější kyselina v plynové fázi., Toto experimentální pozorování naznačují, než alkylové skupiny může stabilizovat anionty i kationty a že solvatační hraje důležitou roli při určování relativní acidities. Proto je nutné slovo opatrnosti. Relativní acidity, na kterých jsou zobecnění uvedená v této kapitole založena, byly stanoveny ve vodě. V plynové fázi jsou často vidět zvraty v pořadí příbuzných sloučenin.“(Eğe, 1999, s. 107)
některé alkylové substituční účinky byly často vysvětleny v učebnicích protichůdnými nebo záhadnými způsoby. , Tak, chemický posun, rozdíly mezi CH3 a CH2 skupiny jsou připisovány v Hornback je kniha k tomu, že „uhlík je mírně více elektronegativní než vodík“ (Hornback, 2006, str. 549) navzdory alkyl skupina byla dříve klasifikována jako slabá induktivní elektron-darování substituent (Hornback, 2006, str. 117). V Vollhardt je učebnice, vztah mezi methylovou skupinu chemických posunů pro řadu CH3X sloučeniny a X elektronegativita je znázorněn v tabulce chybí položka pro X=methyl (Vollhardt & Schore, 2014, str., 389), čímž se zabrání nepohodlnému problému s uhlíkem.
zde ukazuji, že alkylová skupina se chová jako substituent a –i+R. Ačkoli některé faktory (jako jsou pole, sterické nebo solventní účinky) jsou v tomto přístupu implicitně ignorovány, lze tak snadno popsat mnoho aktuálně dostupných teoretických a experimentálních důkazů.
Cδ–Hδ+ bond polarizace bylo experimentálně pozorováno, pro metan (Lazzeretti, Zanasi, & Raynes, 1987), v souladu s větší elektronegativitě uhlíku v poměru k vodíku, 2.55 vs. 2.,20 v Paulingově stupnici (Allred, 1961). Taková polarizace vzorec umožňuje předpovídat dipólový moment směr jednoduché uhlovodíky prostřednictvím aditivní modely, kvantitativní dohody je obvykle skromný (2-methylpropan: 0.3 D odhaduje vs. 0.132 D experimentální) (Dean, 1999).
Protože vodík je použit jako standard v Ingold je klasifikace substituentů (Krygowski & St??pień, 2005), alkylová skupina by měla být klasifikována jako substituent a-I (odtud σ elektronová stahovací skupina). Taková role je znázorněna na obr., 1 pro σ dluhopisů polarizace od toho, co atom Y methylová skupina, i když zadní dluhopisů polarizace se předpokládá, když Y je více elektronegativní než uhlík (např. chlór).
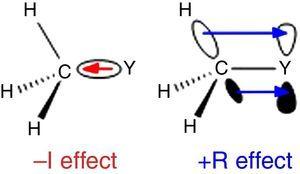
– i (vlevo) a +R (vpravo) účinky methylové skupiny vázané na atom y.
odlišné chování je nalézt pro alkylové skupiny, když připojený k sp2 nebo sp-hybridizované atomy vzhledem k elektronové hustoty dar z alkyl-C–H nebo C–C σ dluhopisů do prázdné p orbital přilehlé atom (nejjednodušší π-systému), jak je znázorněno na Obr. 1. Snížení kyselosti plynné fáze fenolu a kyseliny benzoové substitucí p-methyl (McMahon & Kebarle, 1977)lze tedy připsat pouze významnému efektu π-dárce pro methylovou substituent (ve skutečnosti větší než u methoxy skupiny)., Alkylová skupina by však měla být považována za atypický substituent π-dárce kvůli nedostatku osamělých elektronových párů. Takové σ-bond/π-systém interakce, pojmenované jako hyperconjugation (Mullins, 2012) lze snadno vysvětlit pomocí analogie s π-donor chování lone-pair-ložiska atom (např. chlór) pro prázdné p orbital, když C–C nebo C–H vazby (spíše než elektron osamělé páry) alkylové skupiny jsou zapojeny jako elektron-uvolnění jednotky v hyperconjugative interakce., Zajímavé je, π→σ* interakce (negativní hyperconjugation) jsou obvykle zanedbatelné pro alkylové skupiny chybí elektronegativní atomy (Bocca, Pontes, & Basso, 2004).
některé molekulární strukturální rysy lze racionalizovat na základě vlastností alkylové skupiny. Například větší délky vazby CO nalezené v methylketonech (aceton: exp. 1.210 Å, calc. 1.193 Å) ve srovnání s příbuznými aldehydy (acetaldehyd: exp . 1.209 Å, calc. 1.,188Å) (Berry, Waltman, Pacansky, & Hagler, 1995), lze přičíst k stabilizaci zwitterionic rezonance formulář (viz Obr. 2) prostřednictvím alkylové skupiny π-darování atomu karbonylového uhlíku, čímž se oslabuje dvojná vazba karbonylové skupiny.
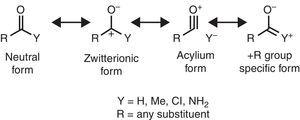
neutrální (vlevo) a zwitterionic (vpravo) rezonanční formy karbonylové sloučeniny.
Hyperconjugative interakce jsou závislé na uspořádání C–H (nebo C–C) dluhopisy vzhledem k p orbital přilehlé atom Y, nejvíce efektivní interakce odpovídající téměř paralelní uspořádání. Například toluen Csp3–H vazba téměř kolmo k rámcové letadlo je o něco delší než ostatní Csp3–H vazby (0.002 Å, Hameka & Jensen, 1996)., Geometrie závislost hyperconjugation umožňuje vysvětlit konformační analýzu methyl substituovaných nenasycených sloučenin, jako propen (Liberles, O ‚ leary, Eilers, & Whitman, 1972) nebo acetaldehyd (Muñoz-Caro, Niño, & Moule, 1994).
Jako známý důsledek π-donor chování alkylovou skupinu, alkyl substituce výnosy více electron-rich alkenů a opera (Libit & Hoffmann, 1974)., Vysoká reaktivita z alkyl-substituovaných areně v SEAr reakce tak může být připsána na stabilizaci odpovídající Wheland intermediate prostřednictvím π-elektronů darování.
–I+R chování alkyl skupiny umožňuje vysvětlit řadu funkcí, z alkyl-substituovaných sloučenin, jako jsou dipólové momenty, spektroskopické vlastnosti a reaktivita (v plynné fázi a v roztoku média), jak je uvedeno níže.
dipólové momenty
chování alkylové skupiny v alifatických sloučeninách se odráží také v dipólových okamžicích., Vektory dipólového momentu pro propan a 2-methylpropan (Tasi et al., 1997), stejně jako některé substituované bicyclooctanes (Böhm & Exner, 2004), lze přičíst k odnímání efekt (–I) methylové skupiny ve srovnání s vodíkem (viz Obr. 3).
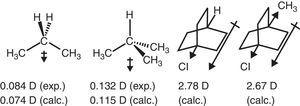
dipólové momenty propanu a substituovaných bicyklooktanů.
V kontrastu, π-donor charakter methyl skupiny (+R) je nezbytné vysvětlit zvýšení dipólového momenty nitrobenzen a benzonitrile prostřednictvím p-methyl substituce (Hnědá, 1959) (viz Obr. 4).
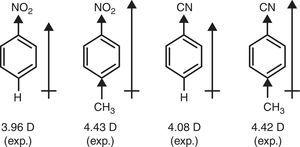
dipólové momenty benzenových derivátů.
Molekulární dipólové momenty lze spolehlivě vypočítat pomocí současných výpočetních metod., Zajímavé je, že vypočtené dipólové momentové vektory pro sadu jednoduchých uhlovodíků (Tasi et al., 1997) umožnily vyvozovat dvojí roli pro methylovou skupinu: elektron-odnímání, když připojený k sp3 atomy uhlíku, ale elektron-darování, když povinen sp2 nebo sp3 uhlíků.
takové duální chování alkylového substituentu je také pozorováno u heteroatomových sloučenin. Postupný pokles dipólového momentu je tedy pozorován u po sobě jdoucích methylových substitucí na amoniaku (NH3, 1,47 D; MeNH2, 1,31 D; Me2NH, 1,01 D; Me3N, 0.,61 D) (Le Févre & Russell, 1947), v dohodě s progresivní snížení dusíku elektronové hustoty (Hehre & Pople, 1970). V kontrastu, dipólový moment vylepšení (od 1.53 D 1.68 D) (Nelson, Lide, & Maryott, 1967) se nachází na N,N-dimethyl substituce na anilin (Targema, Obi-Egbedi, & Adeoye, 2013), důsledně se zvýší z π-donor znak pro amino skupinu (Hinchliffe & Kidd, 1980), vzhledem k +R příspěvků methylové substituenty.,
Spektroskopické vlastnosti
Spektroskopické vlastnosti mnoha organických sloučenin lze snadno racionalizovat za předpokladu chování a-i+R pro alkylovou skupinu jako obecný rys. Chemický posun atomu NMR lze tedy považovat za experimentální měřítko hustoty elektronů v odpovídající poloze jádra, i když mohou být zapojeny i jiné účinky – jako jsou anizotropní magnetická pole. Hřiště posuny vyvolané methyl substituent na sp3 uhlíkových atomů (+9.6 ppm, v 13C NMR) nebo odpovídající vázané atomy vodíku (+0.,63ppm v 1H NMR) (Pretsch, Bühlmann, & Badertscher, 2009) jsou konzistentní s chování typické –já skupin (jako jsou halogenové atomy).
Alkyl substituční efekty na NMR chemické posuny alkenů show elektronovou hustotu pokles v α poloze (+12.9 ppm pro 13C NMR; +0.45 ppm pro 1H NMR), stejně jako hustota zvýšit v β poloze (-7.4 ppm pro 13C; -0.31/-0.40 ppm pro 1H), důsledně s –I+R efektu, i když anizotropní efekty (jako prsten proudy), může také hrát roli. Takové chování a-i+R se také vyskytuje u alkynů podle 13C NMR spektroskopie (+8.,5ppm pro α pozici, -3,6 ppm pro β pozici).
dichotomické chování alkylových substituentů na π-systémech (zvýšení hustoty elektronů pro Atom α, snížení hustoty elektronů pro Atom β) nelze vysvětlit na základě jednoduchého chování (například efekt +i).
–I+R chování (Meier, 2007) je pozorován přes 15N NMR spektroskopie pro alkyl substituce na aminy a amidy v závislosti na dusíku hybridizace (hřiště směny pro alifatické aminy, upfield směny pro Nsp2-ložiska sloučeniny – jako anilines a amidy).,
spojovací konstanty NMR jsou také závislé na substituentních elektronických vlastnostech (stejně jako na některých geometrických vlastnostech). Tím, významný pokles je nalézt pro 1H–1H tažné konstanty prostřednictvím methyl substituce (trans, -2.3 Hz; cis, -1.6 Hz; gem, -0.4 Hz), v kvalitativní shodě s údaji z typických elektron-odnímání skupin, jako je atom fluoru (trans, -6.3 Hz; cis, -6.9 Hz; gem, -5.7 Hz). Pozitivní přínos pro methylovou substituci na 13C-1h spojovacích konstantách alifatických sloučenin (+1.,0Hz), je také kvalitativně konzistentní s těmi z jiných –i skupin (fluor, +24Hz).
Infračervené spektroskopie je také citlivý na substituentu vlastnosti, jak je znázorněno tím, CO strečink frekvence karbonylové sloučeniny jako funkce odpovídající substituent Y, který může být racionalizováno z hlediska rezonance forem (Obr. 2). Užíváním alifatického aldehydu (ca., 1725cm–1) jako referenční, rudý posuv (snížení vlnočtu) vyvolané +jsem substituent (acetyltrimethylsilane, 1645cm–1: Soderquist & Hsu, 1982), lze přičíst k stabilizaci zwitterionic formu. Místo toho, blueshift vyvolané jsem substituent (acyl chloridy, >1800cm–1: Pretsch et al., 2009) lze vysvětlit pomocí dvou alternativních nebo souběžných mechanismů (destabilizace zwitterionické formy a/nebo příspěvek acyliové iontové formy). Konečně, redshifts vyvolané + R substituenty (amides, ca., 1680cm-1: Pretsch et al., 2009) lze připsat příspěvku specifické rezonanční formy. Mírný redshift indukovaný alkylovou skupinou (methyl ketony, ca. 1715cm–1) vykazuje čistý efekt darování elektronů (tedy převahu +R efektu over –I vlastností). Čistý dárce účinku karbonylové skupině vázané alkylové skupiny je v souladu s větší dipólový moment acetonu (2.88 D) vzhledem k formaldehydu (2.33 D) (Nelson et al., 1967).
vliv alkylové skupiny na UV-VIS spektra mnoha sloučenin lze také vysvětlit z hlediska elektronických efektů., Tak, bathochromic posuny vyvolané alkylové skupiny na UV absorpční pásma α,β-nenasycené sloučeniny (+10nm v α poloze, +12nm v β poloze), konjugované polyeny (+5 nm) nebo deriváty benzenu (+3,0 nm) jsou kvalitativně konzistentní s účinky typické π-donor skupiny (např. chlór).
acidobazická reaktivita plynné fáze
relativní bazicita alifatických aminů ve vodném roztoku byla přičítána předpokládanému účinku alkylové skupiny + i (Sorrell, 2006)., Interestingly, the irregular basicity order of amines in water (Me2NH>MeNH2>Me3N>NH3, as shown by the pKa values for the corresponding conjugated acids: 10.77>10.62>9.80>9.,246) (Dean, 1999) je znečištěné rozpouštědlo účinků, jak dokládá systematické basicity, aby aminů v plynné fázi (Me3N>Me2NH>MeNH2>NH3) (Brauman, Riveros, & Blair, 1971)., Ačkoli plynné fázi basicity objednat lze připsat obvykle předpokládá, +jsem alkyl efekt (Carter, 2007), snížení dusíku elektronové hustoty prostřednictvím methyl střídání bylo skutečně pozorováno pomocí Molekulární Elektrostatický Potenciál výpočty (Baeten, De Proft, & Geerlings, 1995), což naznačuje –I chování pro methylovou skupinu., Vlastně plynné fázi basicity, aby z alifatických aminů je třeba přičíst zvýšení stabilizace substituované amonné ionty, díky alkylové skupině polarizability (Aue, Webb, & Bowers, 1976).
Vzhledem acidities alkoholů ve vodném roztoku (H2O>MeOH>EtOH>iPrOH>tBuOH) byly také připisovány v některých učebnicích se předpokládat, alkyl +I efekt (Johnson, 1999; Šalamounovy ostrovy, Fryhle, & Snyder, 2016)., Jelikož reverzní kyselost cílem je nalézt v plynné fázi, relativní acidities alkoholů ve vodě by měla být připsána na spodní veličiny solvatační enthalpies pro větší alkoxide anionty (Brauman & Blair, 1969).
diskuse o elektronických vlastnostech alkylové skupiny lze také aplikovat na karbaniony. To znamená, že ‚učebnice‘ stabilita pořadí pro jednoduché carbanions (methyl>ethyl>isopropyl>terc-butyl) byl přičíst k předpokládané +I indukční efekt alkylových skupin (Burrows et al.,, 2013; Chaloner, 2015; Roos & Roos, 2014; Smith, 2013). Nicméně, nepravidelný cílem je nalézt pro plynné fázi carbanion stabilities (tBu>>iPr>Et), po dohodě s souběh dvou proti alkyl účinky (DePuy et al., 1989): stabilizační mechanismus, alkyl polarizability (to znamená, že n→σ* hyperconjugation) a destabilizující trend (v souladu s +R roli, za předpokladu, že p-jako chování uhlíku osamělý pár).,
stabilita ostatních reakčních meziproduktů může být také hodnocena na základě účinků alkylové skupiny. To znamená, že dobře známé stabilita pořadí pro carbocations (terciární>střední>primární>methyl) byla někdy připisována pozitivní indukční efekt (Chaloner, 2015; Roos & Roos, 2014)., Zajímavé je, hyperconjugation je prezentována v mnoha učebnicích jako alternativní vysvětlení pro stabilitu pořadí carbocations (Hnědá, Iverson, Anslyn, & Foote, 2013; Burrows et al., 2013), i když obvykle nejednoznačné psaní zabraňuje zjištění, zda jsou obě vysvětlení odpovídají buď dva různé popisy stejného jevu nebo dvou souběžných mechanismů hrát ve stejném směru., Každopádně, stabilita pořadí pro carbocations by měla být připsána na hyperconjugation (tedy+R chování volný p orbital, nejjednodušší π systém), i když další interakce (jako alkyl polarizability) jsou také zapojeny (Aue, 2011).
volné radikály vykazují stejné pořadí stability jako karbokace, což naznačuje stabilizaci alkylovou substitucí. I když takové stability, aby může být odůvodněno na základě předpokládané +I chování, k +R účinek může být případně považován, obdobně ke stabilizaci volných radikálů pomocí elektronové páry nesoucí atomy (Zipse, 2006).,
Reaktivita v roztoku
Vzhledem acidities jednoduchých karboxylových kyselin ve vodném roztoku (kyselina octová>propionová kyselina>kyselina máselná) byly použity v některých učebnicích pro ilustraci předpokládá, +I efektu alkylu skupiny (Sorrell, 2006). Je zajímavé, že v opačném pořadí, je našel, když enthalpies místo toho jsou považovány (Christensen, Izatt, & Hansen, 1967), což naznačuje, že kyselost, aby ve vodném roztoku by měla být připsána na hydrataci entropies., To znamená, že významná mřížka pořadí kapalné vody (odpařování entropie rovnající se 118,89 Jmol–1k–1, na rozdíl od typických hodnot ca. 88Jmol-1k-1 pro většinu kapalin, Dean, 1999) může zavést značné změny v reakční energetice. Zejména, hydrataci apolar molekuly (nebo skupiny), vede k dalšímu rozpouštědla mříž objednání (Blokzijl & Engberts, 1993). V důsledku toho, alkylovou skupinu indukční účinky z experimentálních dat ve vodném roztoku jsou často maskované hydrataci entropies (Calder & Barton, 1971)., Relativní acidities jednoduchých karboxylových kyselin v plynné fázi (Yamdagni & Kebarle, 1973) a acetonitril (Eckert et al., 2009) jsou v souladu s hlavní rolí hydratačních entropií.
nižší kyselost pivalic kyseliny v porovnání s kyselinou octovou, obvykle přičítán předpokládat +I efektu alkylu skupiny (Smith, 2008), je obrácen, když reakce enthalpies jsou považovány za (Eckert et al., 2009).,
předpokládaný účinek alkylové skupiny + i na kyselost jednoduchých karboxylových kyselin ve vodném roztoku lze tedy připsat artefaktu odvozenému od účinků rozpouštědla. Zatímco zvýšení objemu neutrálních rozpuštěných látek vede ke zvýšení hydratace entropie, u iontových druhů se objevuje opačný vztah (Graziano, 2009). V důsledku toho, substituce alkyl (prostřednictvím zvýšení molekulární objemu) vede k stabilizaci (gibbsovy volné energie týče) non-ionizované kyseliny ve vodě, stejně jako destabilizaci odpovídající karboxylový anion, tedy výsledná kyselost sníží.,
větší kyselosti kyselina mravenčí ve srovnání s octové ve vodném roztoku (hodnoty pKa: 3.751 a 4.756, respektive, Dean, 1999) byly rovněž projednány v mnoha učebnicích jako příklad použití indukční účinky (Hart, Hadad, Crainovi, & Hart, 2012; Hornback, 2006; Okuyama & Maskill, 2014; Roos & Roos, 2014). Protože velmi podobné reakční entalpie se podílejí na disociačních reakcích kyseliny mravenčí a kyseliny octové (Christensen et al.,, 1967), větší kyselost kyseliny mravenčí musí být skutečně přičítána hydratačním rozdílům entropie.
Závěry
jasné pochopení indukční a rezonanční efekty je hlavním klíčem pro zdravé učení Organické Chemie (Mullins, 2008). Překvapivě byla téměř všudypřítomná alkylová skupina nesprávně prezentována v mnoha učebnicích jako skupina σ-dárce (+i). Duální chování je však prokázáno alkylovými substituenty v závislosti na hybridizaci sousedního atomu., Tak, alkylové skupiny vázané na alifatické řetězce se chovají jako σ-akceptory (–I, v souladu s větší elektronegativitě uhlíku v poměru k vodíku), vzhledem k tomu, že jsou připojené k π-systémy působit jako π-donorů (+R, vzhledem k hyperconjugative interakce). Řada experimentálních a teoretických dat (dipólové momenty, NMR, ir a UV spektra, reaktivita) souhlasí s takovým dvojím chováním.,
celý analýzu všech dat za tu umožňuje odvozovat malé –I efekt, stejně jako významné +R chování pro alkylové skupiny jako funkce platné ve všech diskusích na spektroskopické a reaktivita vlastnosti organických sloučenin.
střet zájmů
autor deklaruje žádný střet zájmů.