Bowtie indexa el genoma de referencia utilizando un esquema basado en la transformación de Burrows-Wheeler (BWT) y el índice FM . Un índice Bowtie para el genoma humano cabe en 2,2 GB en el disco y tiene una huella de memoria de tan solo 1,3 GB en el momento de la alineación, lo que permite que se consulte en una estación de trabajo con menos de 2 GB de RAM.,
el método común para buscar en un índice FM es el algoritmo de coincidencia exacta de Ferragina y Manzini . Bowtie no simplemente adopta este algoritmo porque la coincidencia exacta no permite errores de secuenciación o variaciones genéticas. Presentamos dos nuevas extensiones que hacen que la técnica sea aplicable a la alineación de lectura corta: un algoritmo de backtracking consciente de la calidad que permite desajustes y favorece alineamientos de alta calidad; y’ doble indexación’, una estrategia para evitar el backtracking excesivo., El alineador de pajarita sigue una política similar a la de Maq, ya que permite un pequeño número de desajustes dentro del final de alta calidad de cada lectura, y coloca un límite superior en la suma de los valores de calidad en posiciones de alineación no coincidentes.
Burrows-Wheeler indexing
El BWT es una permutación reversible de los caracteres en un texto. Aunque originalmente se desarrolló en el contexto de la compresión de datos, la indexación basada en BWT permite buscar textos grandes de manera eficiente en una pequeña huella de memoria., Se ha aplicado a aplicaciones bioinformáticas, incluyendo Conteo de oligómeros , alineación de genoma completo , diseño de sonda de microarray de mosaico y alineación Smith-Waterman a una referencia de tamaño humano .
La transformación Burrows-Wheeler de un texto T, BWT (T), se construye de la siguiente manera. El carácter $ se añade A T, donde $ no está en T y es lexicográficamente menor que todos los caracteres en T. La matriz de Burrows-Wheeler de T se construye como la matriz cuyas filas comprenden todas las rotaciones cíclicas de T.. Las filas se ordenan lexicográficamente., BWT (T) es la secuencia de caracteres en la columna más a la derecha de la matriz Burrows-Wheeler (figura 1a). BWT(T) tiene la misma longitud que el texto original T.
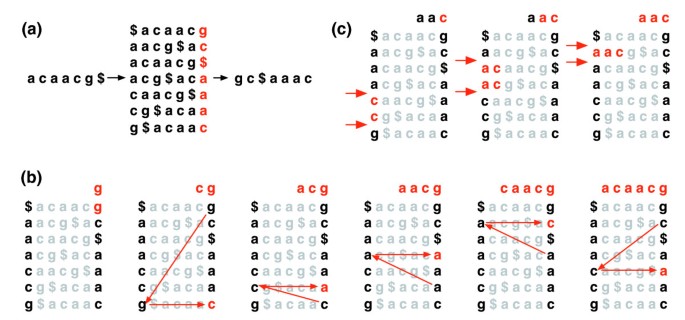
de Burrows-Wheeler transformar. (a) la matriz Burrows-Wheeler y transformación para «acaacg». B) Las medidas adoptadas por EXACTMATCH para identificar el rango de filas y, por lo tanto, el conjunto de sufijos de referencia, precedidos por «aac»., (c) UNPERMUTE aplica repetidamente la última primera asignación (LF) para recuperar el texto original (en rojo en la línea superior) de la transformación de Burrows-Wheeler (en negro en la columna de la derecha).
Esta matriz tiene una propiedad llamada ‘apellido (LF) el mapeo. La i-ésima aparición del carácter X en la última columna corresponde al mismo carácter de texto que la i-ésima aparición de X en la primera columna. Esta propiedad subyace a los algoritmos que utilizan el índice BWT para navegar o buscar el texto., La figura 1B ilustra UNPERMUTE, un algoritmo que aplica la asignación LF repetidamente para recrear T A partir de BWT(T).
la asignación LF también se utiliza en la coincidencia exacta. Debido a que la matriz se ordena lexicográficamente, las filas que comienzan con una secuencia dada aparecen consecutivamente. En una serie de pasos, el algoritmo EXACTMATCH (figura 1C) calcula el rango de filas de matriz que comienzan con sufijos sucesivamente Más largos de la consulta. En cada paso, el tamaño del rango se reduce o permanece igual., Cuando el algoritmo se completa, las filas que comienzan con S0 (la consulta completa) corresponden a las ocurrencias exactas de la consulta en el texto. Si el rango está vacío, el texto no contiene la consulta. UNPERMUTE es atribuible a Burrows y Wheeler y EXACTMATCH a Ferragina y Manzini . Vea el archivo de datos adicionales 1 (discusión suplementaria 1) para más detalles.
la búsqueda de alineaciones inexactas
EXACTMATCH es insuficiente para una alineación de lectura corta porque las alineaciones pueden contener desajustes, que pueden deberse a errores de secuenciación, diferencias genuinas entre los organismos de referencia y consulta, o ambos., Presentamos un algoritmo de alineación que realiza una búsqueda de retroceso para encontrar rápidamente alineaciones que satisfagan una política de alineación especificada. Cada carácter en una lectura tiene un valor de calidad numérico, con valores más bajos que indican una mayor probabilidad de un error de secuenciación. Nuestra Política de alineación permite un número limitado de desajustes y prefiere alineaciones donde la suma de los valores de calidad en todas las posiciones no coincidentes es baja.
la búsqueda procede de manera similar a EXACTMATCH, calculando rangos de matriz para sufijos de consulta sucesivamente Más largos., Si el rango se vuelve vacío (no aparece un Sufijo en el texto), entonces el algoritmo puede seleccionar una posición de consulta ya coincidente y sustituir una base diferente allí, introduciendo un desajuste en la alineación. La búsqueda EXACTMATCH se reanuda justo después de la posición sustituida. El algoritmo selecciona solo aquellas sustituciones que son consistentes con la política de alineación y que producen un sufijo modificado que aparece al menos una vez en el texto. Si hay varias posiciones de sustitución candidatas, entonces el algoritmo selecciona con avidez una posición con un valor de calidad mínimo.,
los escenarios de retroceso se desarrollan dentro del contexto de una estructura de pila que crece cuando se introduce una nueva sustitución y se reduce cuando el alineador rechaza todas las alineaciones candidatas para las sustituciones actualmente en la pila. Vea la Figura 2 para una ilustración de cómo podría proceder la búsqueda.
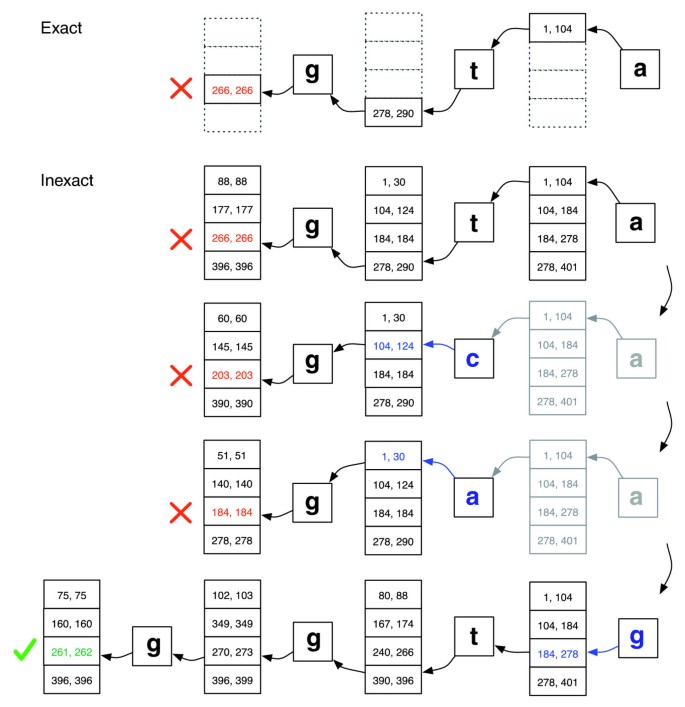
correspondencia Exacta frente a inexactas que realice la alineación., Ilustración de cómo se procede EXACTMATCH (arriba) y el alineador de Bowtie (abajo) cuando no hay una coincidencia exacta para la consulta ‘ggta’ pero hay una alineación de un desajuste cuando ‘a’ se reemplaza por ‘g’. Los pares de números en caja denotan rangos de filas de matriz que comienzan con el sufijo observado hasta ese punto. Una X roja marca donde el algoritmo encuentra un rango vacío y aborta (como en EXACTMATCH) o retrocede (como en el algoritmo inexact). Una marca de verificación verde donde el algoritmo encuentra un rango no vacío que delimita una o más ocurrencias de una alineación reportable para la consulta.,
En resumen, Bowtie lleva a cabo una búsqueda de calidad, codiciosa, aleatoria y en profundidad a través del espacio de posibles alineaciones. Si existe una alineación válida, entonces Bowtie lo encontrará (sujeto al techo de retroceso discutido en la siguiente sección). Debido a que la búsqueda es codiciosa, la primera alineación válida encontrada por Bowtie no necesariamente será la ‘mejor’ en términos de número de desajustes o en términos de calidad., El usuario puede indicar a Bowtie que continúe buscando hasta que pueda probar que cualquier alineación que informe es ‘mejor’ en términos de número de desajustes (utilizando la opción best best). En nuestra experiencia, este modo es dos o tres veces más lento que el modo predeterminado. Esperamos que el modo predeterminado más rápido sea el preferido para grandes proyectos de re-secuenciación.
El usuario también puede optar por Bowtie para reportar todas las alineaciones hasta un número especificado (opción-k) o todas las alineaciones sin límite en el número (opción-a) para una lectura dada., Si en el curso de su búsqueda Bowtie encuentra N alineaciones posibles para un conjunto dado de sustituciones, pero el Usuario ha solicitado solo alineaciones k donde K < N, Bowtie reportará K de las alineaciones n seleccionadas al azar. Tenga en cuenta que estos modos pueden ser mucho más lentos que el predeterminado. En nuestra experiencia, por ejemplo,- k 1 es más del doble de rápido que-k 2.
retroceso excesivo
el alineador descrito hasta ahora puede, en algunos casos, encontrar secuencias que causan retroceso excesivo., Esto ocurre cuando el alineador pasa la mayor parte de su esfuerzo infructuosamente retrocediendo a posiciones cercanas al final de 3′ de la consulta. Bowtie mitiga el retroceso excesivo con la novedosa técnica de ‘doble indexación’. Se crean dos índices del genoma: uno que contiene el BWT del genoma, llamado índice’ forward’, y un segundo que contiene el BWT del genoma con su secuencia de caracteres invertida (no complementada reversa) llamada índice ‘mirror’. Para ver cómo ayuda esto, considere una política de coincidencia que permita una discrepancia en la alineación., Una alineación válida con un desajuste cae en uno de los dos casos según los cuales la mitad de la lectura contiene el desajuste. Bowtie procede en dos fases correspondientes a esos dos casos. La fase 1 carga el índice hacia adelante en la memoria e invoca al alineador con la restricción que no puede sustituir en las posiciones de la mitad derecha de la consulta. La fase 2 utiliza el índice mirror e invoca al alineador en la consulta invertida, con la restricción de que el alineador no puede sustituir en las posiciones de la mitad derecha de la consulta invertida (la mitad izquierda de la consulta original)., Las restricciones en el retroceso en la mitad derecha evitan el retroceso excesivo, mientras que el uso de dos fases y dos índices mantiene la sensibilidad completa.
Desafortunadamente, no es posible evitar un retroceso excesivo cuando se permite que las alineaciones tengan dos o más desajustes. En nuestros experimentos, hemos observado que el retroceso excesivo es significativo solo cuando una lectura tiene muchas posiciones de baja calidad y no se alinea o se alinea mal con la referencia., Estos casos pueden desencadenar más de 200 retrocesos por Lectura porque hay muchas combinaciones legales de posiciones de baja calidad que deben explorarse antes de que se agoten todas las posibilidades. Mitigamos este costo imponiendo un límite en el número de retrotracks permitidos antes de que se termine una búsqueda (predeterminado: 125). El límite evita que se reporten algunas alineaciones legítimas y de baja calidad, pero esperamos que esto sea una compensación deseable para la mayoría de las aplicaciones.,
Phased MAQ-like search
Bowtie permite al usuario seleccionar el número de desajustes permitidos (por defecto: dos) en el final de alta calidad de una lectura (por defecto: las primeras 28 bases), así como la distancia máxima de calidad aceptable de la alineación general (por defecto: 70). Se asume que los valores de calidad siguen la definición en PHRED, donde p es la probabilidad de error y Q = -10log p.
tanto el complemento leído como su complemento inverso son candidatos para la alineación con la referencia. Para mayor claridad, esta discusión considera solo la orientación hacia el futuro., Vea el archivo de datos adicionales 1 (discusión suplementaria 2) para una explicación de cómo se incorpora el complemento inverso.
las primeras 28 bases en el extremo de alta calidad de la lectura se denominan la «semilla». La semilla consta de dos mitades: el 14 pb en el extremo de alta calidad (generalmente el extremo de 5′) y el 14 pb en el extremo de baja calidad, denominados ‘hi-half’ y ‘lo-half’, respectivamente., Asumiendo la política predeterminada (dos desajustes permitidos en la semilla), una alineación reportable caerá en uno de cuatro casos: sin desajustes en semilla (caso 1); sin desajustes en hi-half, uno o dos desajustes en lo-half (caso 2); sin desajustes en lo-half, uno o dos desajustes en hi-half (caso 3); y un desajuste en hi-half, un desajuste en lo-half (caso 4).
todos los casos permiten cualquier número de desajustes en la parte no sembrada de la lectura y todos los casos también están sujetos a la restricción de distancia de calidad.,
el algoritmo Bowtie consta de tres fases que alternan entre el uso de los índices de avance y espejo, como se ilustra en la Figura 3. La fase 1 utiliza el índice del espejo e invoca al alineador para encontrar alineaciones para los casos 1 y 2. Las fases 2 y 3 cooperan para encontrar alineaciones para el caso 3: La Fase 2 Encuentra alineaciones parciales con desajustes solo en el hi-half y la fase 3 intenta extender esas alineaciones parciales a alineaciones completas. Finalmente, la fase 3 invoca al alineador para encontrar alineaciones para el caso 4.,

Las tres fases de la Pajarita algoritmo para la Maq-como la política. Un enfoque de tres fases encuentra alineaciones para casos de dos desajustes 1 a 4 al tiempo que minimiza el retroceso. La fase 1 utiliza el índice del espejo e invoca al alineador para encontrar alineaciones para los casos 1 y 2. Las fases 2 y 3 cooperan para encontrar alineaciones para el caso 3: La Fase 2 Encuentra alineaciones parciales con desajustes solo en el hi-half, y la fase 3 intenta extender esas alineaciones parciales a alineaciones completas., Finalmente, la fase 3 invoca al alineador para encontrar alineaciones para el caso 4.
resultados de rendimiento
evaluamos el rendimiento de la pajarita utilizando lecturas del proyecto piloto de 1.000 genomas (archivo de lectura corta del Centro Nacional de Información Biotecnológica:SRR001115). Un total de 8,84 millones de lecturas, aproximadamente un carril de datos de un instrumento Illumina, se recortaron a 35 PB y se alinearon con el genoma humano de referencia . A menos que se especifique lo contrario, los datos de lectura no se filtran ni modifican (además de recortar) de la forma en que aparecen en el archivo., Esto lleva a que entre el 70% y el 75% de las lecturas se alineen en algún lugar con el genoma. En nuestra experiencia, esto es típico para los datos sin procesar del archivo. Un filtrado más agresivo conduce a mayores tasas de alineación y una alineación más rápida.
todas las ejecuciones se realizaron en una sola CPU. Las aceleraciones de Bowtie se calcularon como una relación de los tiempos de alineación del reloj de pared. Tanto el reloj de pared como los tiempos de CPU se dan para demostrar que la carga de entrada/salida y la contención de la CPU no son factores significativos.
el tiempo requerido para construir el índice de la pajarita no se incluyó en los tiempos de ejecución de la pajarita., A diferencia de las herramientas de la competencia, Bowtie puede reutilizar un índice pre-calculado para el genoma de referencia a través de Muchas corridas de alineación. Anticipamos que la mayoría de los usuarios simplemente descargarán dichos índices de un repositorio público. The Bowtie site provides indices for current builds of the human, chimpancé, ratón, perro, rata, and Arabidopsis thaliana genomes, as well as many others.
los resultados se obtuvieron en dos plataformas de hardware: una estación de trabajo de escritorio con procesador Intel Core 2 de 2,4 GHz y 2 GB de RAM; y un servidor de gran memoria con un procesador AMD Opteron de cuatro núcleos de 2,4 GHz y 32 GB de RAM., Estos se denotan ‘PC’ y ‘servidor’, respectivamente. Tanto el PC como el servidor ejecutan Red Hat Enterprise Linux como la versión 4.
comparación con SOAP y Maq
Maq es un alineador popular que se encuentra entre las herramientas de código abierto más rápidas de la competencia para alinear millones de lecturas de Illumina al genoma humano. SOAP es otra herramienta de código abierto que se ha reportado y utilizado en proyectos de lectura corta .
La Tabla 1 presenta el rendimiento y la sensibilidad de Bowtie v0.9.6, SOAP v1.10 y Maq v0.6. 6. SOAP no se pudo ejecutar en el PC porque la huella de memoria de SOAP excede la memoria física del PC. El jabón.,se utilizó la versión de contig del binario SOAP. Para la comparación con SOAP, Bowtie se invocó con ‘-V 2’ para imitar la política de coincidencia predeterminada de SOAP (que permite hasta dos desajustes en la alineación y no tiene en cuenta los valores de calidad), y con ‘max maxns 5’ para simular la política predeterminada de SOAP de filtrar lecturas con cinco o más bases de no confianza. Para la comparación de Maq, Bowtie se ejecuta con su política predeterminada, que imita la política predeterminada de Maq de permitir hasta dos desajustes en las primeras 28 bases y aplicar un límite general de 70 en la suma de los valores de calidad en todas las posiciones de lectura no coincidentes., Para hacer que la huella de memoria de Bowtie sea más comparable a la de Maq, Bowtie se invoca con la opción ‘-z’ en todos los experimentos para garantizar que solo el índice delantero o espejo sea residente en la memoria a la vez.
el número de lecturas alineadas indica que SOAP (67.3%) y Bowtie-v 2 (67.4%) tienen una sensibilidad comparable. De las lecturas alineadas por jabón o pajarita, 99.7% fueron alineadas por ambos, 0.2% fueron alineadas por pajarita pero no jabón, y 0.1% fueron alineadas por jabón pero no pajarita. El Maq (74,7%) y la pajarita (71,9%) también tienen una sensibilidad aproximadamente comparable, aunque la pajarita se retrasa en un 2,8%., De las lecturas alineadas por Maq o Bowtie, 96.0% fueron alineadas por ambos, 0.1% fueron alineadas por Bowtie pero no Maq, y 3.9% fueron alineadas por Maq pero no Bowtie. De las lecturas mapeadas por Maq pero no Bowtie, casi todas se deben a una flexibilidad en el algoritmo de alineación de Maq que permite que algunas alineaciones tengan tres desajustes en la semilla. El resto de las lecturas mapeadas por Maq pero no Bowtie se deben al techo de Bowtie.
la documentación de Maq menciona que las lecturas que contienen ‘artefactos poly-A’ pueden perjudicar el rendimiento de Maq., La tabla 2 presenta el rendimiento y la sensibilidad de Bowtie y Maq cuando el conjunto de lectura se filtra utilizando el comando ‘catfilter’ de Maq para eliminar los artefactos poly-A. El filtro elimina 438,145 de 8,839,010 lecturas. Otros parámetros experimentales son idénticos a los de los experimentos de la tabla 1, y las mismas observaciones sobre la sensibilidad relativa de Bowtie y Maq se aplican aquí.
longitud de lectura y rendimiento
a medida que mejora la tecnología de secuenciación, las longitudes de lectura están creciendo más allá de los 30-bp a 50-bp que se ven comúnmente en las bases de datos públicas hoy en día., Las lecturas de Bowtie, Maq y SOAP admiten longitudes de hasta 1,024, 63 y 60 PB, respectivamente, y las versiones 0.7.0 y posteriores de Maq admiten longitudes de lectura de hasta 127 PB. La Tabla 3 muestra los resultados de rendimiento cuando las tres herramientas se utilizan cada una para alinear tres conjuntos de lecturas no recortadas de 2 M, un conjunto de 36 bp, un conjunto de 50 bp y un conjunto de 76 bp, con el genoma humano en la plataforma del servidor. Cada conjunto de 2 M se muestrea aleatoriamente de un conjunto más grande (archivo de lectura corta NCBI: SRR003084 para 36-bp, SRR003092 para 50-bp, SRR003196 para 76-bp)., Las lecturas se muestrearon de tal manera que los tres conjuntos de 2 M tienen una tasa de error uniforme por base, calculada a partir de las cualidades Phred por base. Todas las lecturas pasan a través del ‘catfilter’de Maq.
Bowtie se ejecuta tanto en su modo predeterminado como en su modo SOAP ‘-v 2’. Bowtie también se da la opción ‘- z ‘ para asegurar que solo el índice delantero o espejo es residente en la memoria a la vez. Se utilizó Maq v0.7.1 en lugar de Maq V0.6.6 para el conjunto de 76-bp porque v0.6.,6 no puede alinear lecturas de más de 63 PB. El jabón no se ejecutó en el set de 76-bp porque no admite lecturas de más de 60 bp.
los resultados muestran que el algoritmo de Maq escala mejor en general a longitudes de lectura más largas que la pajarita o el jabón. Sin embargo, Bowtie en modo SOAP-like ‘-v 2’ también escala muy bien. Bowtie en su modo predeterminado Maq-como escala bien de 36-bp a 50-bp lee, pero es sustancialmente más lento para 76-bp lee, aunque todavía es más que un orden de magnitud más rápido que Maq.,
Parallel performance
La alineación se puede paralelizar distribuyendo lecturas a través de hilos de búsqueda simultáneos. Bowtie permite al usuario especificar un número deseado de hilos (opción-p); Bowtie luego lanza el número especificado de hilos usando la biblioteca pthreads. Los hilos de pajarita se sincronizan entre sí al obtener lecturas, generar resultados, cambiar entre índices y realizar varias formas de contabilidad global, como marcar una lectura como ‘hecha’., De lo contrario, los subprocesos son libres de operar en paralelo, acelerando sustancialmente la alineación en computadoras con múltiples núcleos de procesador. La imagen de memoria del índice es compartida por todos los subprocesos, por lo que la huella no aumenta sustancialmente cuando se utilizan varios subprocesos. La tabla 4 muestra los resultados de rendimiento para ejecutar Bowtie v0.9. 6 en el servidor de cuatro núcleos con uno, dos y cuatro hilos.,
Index building
Bowtie utiliza un algoritmo de indexación flexible que se puede configurar para intercambiar entre el uso de memoria y el tiempo de ejecución. La tabla 5 ilustra esta compensación al indizar todo el genoma humano de referencia (NCBI build 36.3, contigs). Las ejecuciones se realizaron en la plataforma del servidor. El indexador se ejecutó cuatro veces con diferentes límites superiores en el uso de memoria.,
Los tiempos reportados se comparan favorablemente con los tiempos de alineación de herramientas de la competencia que realizan indexación durante la alineación. Se requieren menos de 5 horas para que Bowtie construya y consulte un índice humano completo con 8,84 millones de lecturas del proyecto 1000 Genome (archivo de lectura corta de NCBI:SRR001115) en un servidor, más de seis veces más rápido que la ejecución equivalente de Maq., La fila inferior ilustra que el indexador Bowtie, con los argumentos apropiados, es lo suficientemente eficiente en memoria como para ejecutarse en una estación de trabajo típica con 2 GB de RAM. El archivo de datos adicional 1 (discusiones suplementarias 3 y 4) explica el algoritmo y el contenido del índice resultante.
el software
Bowtie está escrito en C++ y utiliza la biblioteca SeqAn . El convertidor al formato de asignación Maq utiliza código de Maq.