Bowtie indexa o genoma de referência usando um esquema baseado na transformada de Burrows-Wheeler (BWT) e no índice FM . Um índice de Bowtie para o genoma humano se encaixa em 2,2 GB no disco e tem uma pegada de memória de apenas 1,3 GB no tempo de alinhamento, permitindo que ele seja questionado em uma estação de trabalho com menos de 2 GB de RAM.,
O método comum de pesquisa em um índice FM é o algoritmo de correspondência exata de Ferragina e Manzini . Bowtie não simplesmente adota este algoritmo porque a correspondência exata não permite sequenciamento de erros ou variações genéticas. Introduzimos duas novas extensões que tornam a técnica aplicável ao alinhamento de Leitura curta: um algoritmo de backtracking consciente da qualidade que permite discrepâncias e favorece alinhamentos de alta qualidade; e a “indexação dupla”, uma estratégia para evitar retrocessos excessivos., O Bowtie aligner segue uma política semelhante à do Maq, na medida em que permite um pequeno número de discrepâncias dentro da extremidade de alta qualidade de cada leitura, e coloca um limite superior na soma dos valores de qualidade em posições de alinhamento inadequadas.
Burrows-Wheeler indexing
The BWT is a reversible permutation of the characters in a text. Embora originalmente desenvolvido no contexto da compressão de dados, a indexação baseada em BWT permite que textos grandes sejam pesquisados de forma eficiente em uma pequena pegada de memória., Ele tem sido aplicado a aplicações bioinformáticas, incluindo a contagem de oligômeros , alinhamento do genoma inteiro , desenho de microarray, e alinhamento Smith-Waterman para uma referência de tamanho humano .
a transformação de Burrows-Wheeler de um texto T, BWT (T), é construída da seguinte forma. The character $ is appended to T, where $ is not in T and is lexicograficamente less than all characters in T. The Burrows-Wheeler matrix of T is constructed as the matrix whose rows comprise all cyclic rotations of T$. As linhas são ordenadas lexicograficamente., BWT (T) é a sequência de caracteres na coluna mais à direita da matriz Burrows-Wheeler (figura 1a). BWT(T) tem o mesmo comprimento que o texto original T.
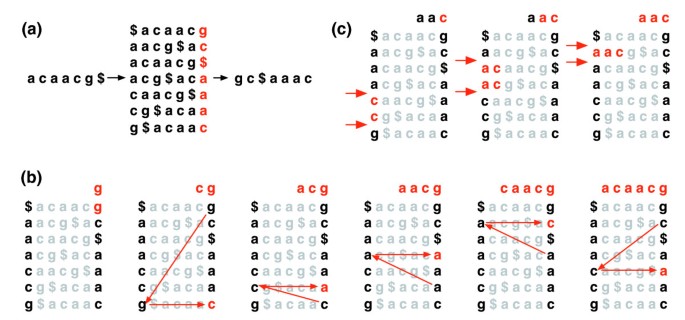
de Burrows-Wheeler transformação. a) a matriz de Burrows-Wheeler e a transformação de “acaacg”. b) As medidas tomadas pela EXACTMATCH para identificar a gama de linhas e, por conseguinte, o conjunto de sufixos de referência, prefixados por “aac”., (c) UNPERMUTE repetidamente aplica o último primeiro mapeamento (LF) para recuperar o texto original (em vermelho na linha superior) da transformada Burrows-Wheeler (em preto na coluna mais à direita).
esta matriz tem uma propriedade chamada ‘last first (LF) mapping’. A i-ésima ocorrência do carácter X na última coluna corresponde ao mesmo carácter de texto que a i-ésima ocorrência de X na primeira coluna. Esta propriedade está subjacente a algoritmos que usam o índice BWT para navegar ou pesquisar o texto., A figura 1b ilustra UNPERMUTE, um algoritmo que aplica o mapeamento LF repetidamente para recriar T a partir de BWT (T).
O mapeamento LF também é usado na correspondência exata. Como a matriz é ordenada lexicograficamente, as linhas que começam com uma dada sequência aparecem consecutivamente. Em uma série de passos, o algoritmo de EXACTMATCH (figura 1c) calcula o intervalo de linhas de matriz começando com sufixos sucessivamente mais longos da consulta. Em cada passo, o tamanho da faixa ou encolhe ou permanece o mesmo., Quando o algoritmo completa, as linhas que começam com S0 (a consulta inteira) correspondem a ocorrências exatas da consulta no texto. Se o intervalo estiver vazio, o texto não contém a consulta. A UNPERMUTE é imputável ao Burrows e ao Wheeler e à EXACTMATCH à Ferragina e ao Manzini . Ver ficheiro de dados adicionais 1 (discussão suplementar 1) para mais pormenores.
A Procura de alinhamentos inexatos
EXACTMATCH é insuficiente para alinhamento de Leitura curta porque alinhamentos podem conter discrepâncias, o que pode ser devido a erros de sequenciação, diferenças genuínas entre organismos de referência e consulta, ou ambos., Introduzimos um algoritmo de alinhamento que conduz uma pesquisa de backtracking para encontrar rapidamente alinhamentos que satisfaçam uma política de alinhamento especificada. Cada caractere em uma leitura tem um valor de qualidade numérica, com valores mais baixos indicando uma maior probabilidade de um erro de sequenciação. Nossa Política de alinhamento permite um número limitado de desfasamentos e prefere alinhamentos onde a soma dos valores de qualidade em todas as posições incompatíveis é baixa.
a pesquisa prossegue similarmente ao EXACTMATCH, calculando intervalos de matriz para sufixos de consulta sucessivamente mais longos., Se o intervalo ficar vazio (um sufixo não ocorre no texto), então o algoritmo pode selecionar uma posição de consulta já emparelhada e substituir uma base diferente lá, introduzindo um desfasamento no alinhamento. A pesquisa de EXACTMATCH recomeça logo após a posição substituída. O algoritmo seleciona apenas as substituições que são consistentes com a Política de alinhamento e que produzem um sufixo modificado que ocorre pelo menos uma vez no texto. Se houver várias posições de substituição candidatas, então o algoritmo seleciona com avidez uma posição com um valor de qualidade mínimo.,
cenários de retrocesso jogam no contexto de uma estrutura de pilha que cresce quando uma nova substituição é introduzida e encolhe quando o aligner rejeita todos os alinhamentos candidatos para as substituições atualmente na pilha. Ver Figura 2 para uma ilustração de como a pesquisa pode prosseguir.
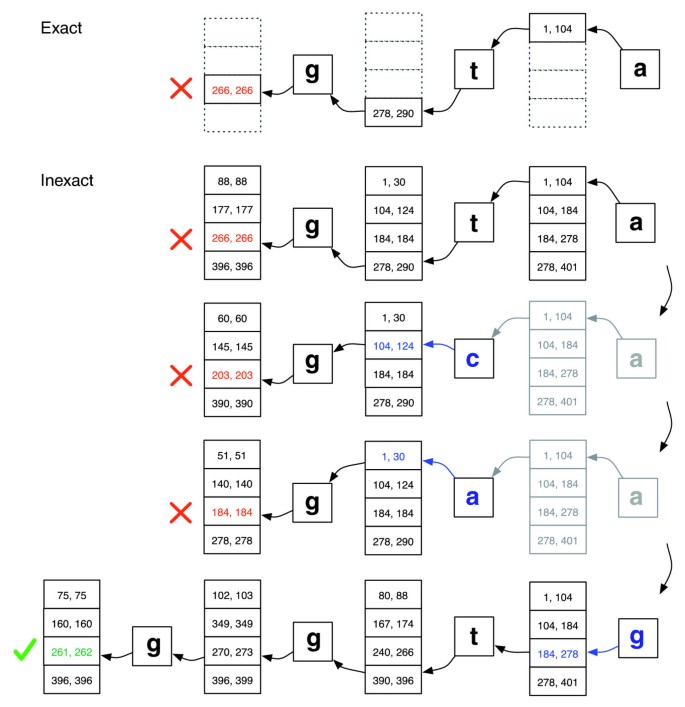
a correspondência Exata versus inexata de alinhamento., Ilustração de como EXACTMATCH (top) e aligner (bottom) de Bowtie procedem quando não existe uma correspondência exata para a consulta “ggta”, mas há um alinhamento unidireccional quando ” a “é substituído por “g”. Pares de números encaixotados denotam intervalos de linhas de matriz começando com o sufixo observado até aquele ponto. A red X marks where the algorithm encounters an empty range and either aborts (as in EXACTMATCH) or backt tracks (as in the inexact algorithm). Uma marcação verde onde o algoritmo encontra um intervalo não vazio que delimita uma ou mais ocorrências de um alinhamento relatável para a consulta.,
Em suma, Bowtie conduz uma pesquisa de qualidade, gananciosa, aleatória, de profundidade através do espaço de possíveis alinhamentos. Se existe um alinhamento válido, então Bowtie vai encontrá-lo (sujeito ao teto backtrack discutido na seção seguinte). Como a busca é gananciosa, o primeiro alinhamento válido encontrado por Bowtie não será necessariamente o “melhor” em termos de número de desajustamentos ou em termos de qualidade., O usuário pode instruir Bowtie para continuar a busca até que ele possa provar que qualquer alinhamento que ele relata é “melhor” em termos de número de desfasamentos (usando a opção –best). Em nossa experiência, este modo é duas a três vezes mais lento do que o modo padrão. Esperamos que o modo padrão mais rápido será preferido para grandes projetos de re-sequenciação.
O usuário também pode optar por Bowtie para relatar todos os alinhamentos até um número especificado (opção-k) ou todos os alinhamentos sem limite no número (opção-a) para uma determinada leitura., Se, no decurso da sua pesquisa, o Bowtie encontrar n possíveis alinhamentos para um determinado conjunto de substituições, mas o utilizador solicitou apenas alinhamentos K onde o K < N, O Bowtie irá reportar K dos alinhamentos n seleccionados aleatoriamente. Note que estes modos podem ser muito mais lentos do que o padrão. Na nossa experiência, por exemplo,- k 1 é mais de duas vezes mais rápido que-k 2.
backtracking excessivo
o aligner, tal como descrito até agora, pode, em alguns casos, encontrar sequências que causam backtracking excessivo., Isto ocorre quando o aligner gasta a maior parte de seu esforço frutificadamente retrocedendo para posições próximas ao fim de 3′ da consulta. Bowtie mitiga o excesso de backtracking com a nova técnica de “dupla indexação”. São criados dois índices do genoma: um contendo o BWT do genoma, chamado de índice ‘forward’, e outro contendo o BWT do genoma com a sua sequência de caracteres invertida (não complementada ao contrário) chamada de índice’ mirror’. Para ver como isso ajuda, considere uma política de correspondência que permite um desfasamento no alinhamento., Um alinhamento válido com um desfasamento cai em um dos dois casos de acordo com o qual metade da leitura contém o desfasamento. A Bowtie prossegue em duas fases correspondentes a esses dois casos. A Fase 1 carrega o índice forward para a memória e invoca o aligner com a restrição que ele pode não substituir em posições na metade direita da consulta. A Fase 2 usa o mirror index e invoca o aligner na consulta invertida, com a restrição de que o aligner não pode substituir em posições na metade direita da consulta invertida (a metade esquerda da consulta original)., As restrições de retrocesso para a metade direita impedem o retrocesso excessivo, ao passo que a utilização de duas fases e dois índices mantém a sensibilidade total.
infelizmente, não é possível evitar um retrocesso excessivo completamente quando os alinhamentos são permitidos ter dois ou mais desajustamentos. Em nossas experiências, temos observado que o excesso de backtracking é significativo apenas quando uma leitura tem muitas posições de baixa qualidade e não se alinha ou se alinha mal à referência., Estes casos podem desencadear mais de 200 backtracks por leitura, porque existem muitas combinações legais de posições de baixa qualidade a serem exploradas antes de todas as possibilidades serem esgotadas. Nós mitigamos este custo, impondo um limite no número de backtracks permitidos antes que uma pesquisa seja terminada (padrão: 125). O limite impede a comunicação de alguns alinhamentos legítimos e de baixa qualidade, mas esperamos que este seja um compromisso desejável para a maioria das aplicações.,
busca faseada igual a Maq
Bowtie permite ao usuário selecionar o número de desfasamentos permitidos (padrão: dois) na extremidade de alta qualidade de uma leitura (padrão: as primeiras 28 bases), bem como a distância de qualidade máxima aceitável do alinhamento global (padrão: 70). Assume-se que os valores de qualidade seguem a definição em PHRED , onde p é a probabilidade de erro e Q = – 10log P.
tanto a leitura como o seu complemento reverso são candidatos ao alinhamento à referência. Por uma questão de clareza, esta discussão considera apenas a orientação para o futuro., Ver ficheiro de dados adicionais 1 (discussão suplementar 2) para uma explicação de como o complemento inverso é incorporado.as primeiras 28 bases na extremidade de alta qualidade da leitura são denominadas “sementes”. A semente consiste em duas metades: a 14 bp na extremidade de alta qualidade (geralmente a extremidade de 5′) e a 14 bp na extremidade de baixa qualidade, denominada “hi-Hal” e “lo-hal”, respectivamente., Assumindo a Política padrão (dois desfasamentos permitidos na semente), um alinhamento relatável cairá em um de quatro casos: Nenhum desfasamento na semente (caso 1); nenhum desfasamento na metade hi, um ou dois desfasamentos na metade lo (caso 2); nenhum desfasamento na metade lo, um ou dois desfasamentos na metade hi (caso 3); e um desfasamento na metade hi, um desfasamento na metade lo (caso 4).
Todos os casos permitem qualquer número de desfasamentos na parte sem necessidade da leitura e todos os casos também estão sujeitos à restrição de distância de qualidade.,
o algoritmo de Bowtie consiste em três fases que alternam entre o uso dos índices forward e mirror, como ilustrado na Figura 3. A Fase 1 usa o mirror index e invoca o aligner para encontrar alinhamentos para os casos 1 e 2. As fases 2 e 3 cooperam para encontrar alinhamentos para o caso 3: A Fase 2 Encontra alinhamentos parciais com desajustamentos apenas na metade III e a fase 3 tenta estender esses alinhamentos parciais em alinhamentos completos. Finalmente, a fase 3 invoca o candidato para encontrar alinhamentos para o caso 4.,

As três fases do Bowtie algoritmo para a Maq-gosto de política. Uma abordagem de três fases encontra alinhamentos para dois casos de desajustamento 1 a 4, minimizando o retrocesso. A Fase 1 usa o mirror index e invoca o aligner para encontrar alinhamentos para os casos 1 e 2. As fases 2 e 3 cooperam para encontrar alinhamentos para o caso 3: A Fase 2 Encontra alinhamentos parciais com desajustamentos apenas na metade III, e a Fase 3 tenta estender esses alinhamentos parciais em alinhamentos completos., Finalmente, a fase 3 invoca o candidato para encontrar alinhamentos para o caso 4.
resultados de desempenho
avaliamos o desempenho de Bowtie usando leituras do piloto do projeto 1000 Genomes (National Center for Biotechnology Information Short Read Archive:SRR001115). Um total de 8,84 milhões de leituras, cerca de uma faixa de dados de um instrumento de iluminação, foram reduzidos para 35 bp e alinhados com o genoma de referência humana . Salvo indicação em contrário, os dados de leitura não são filtrados ou modificados (além de aparar) da forma como aparecem no arquivo., Isto leva a cerca de 70% a 75% das leituras alinhadas em algum lugar ao genoma. Na nossa experiência, isto é típico para os dados brutos do arquivo. Filtragem mais agressiva leva a maiores taxas de alinhamento e alinhamento mais rápido.
Todas as corridas foram realizadas em uma única CPU. Os speedups de Bowtie foram calculados como uma proporção de tempos de alinhamento de parede-relógio. Ambos os tempos de clock de parede e CPU são dados para demonstrar que a carga de entrada/saída e a contenção de CPU não são fatores significativos.
The time required to build the Bowtie index was not included in the Bowtie running times., Ao contrário de ferramentas concorrentes, Bowtie pode reutilizar um índice pré-computado para o genoma de referência em muitas corridas de alinhamento. Prevemos que a maioria dos usuários irá simplesmente baixar tais índices a partir de um repositório público. O site Bowtie fornece índices para as construções atuais do humano, chimpanzé, rato, cão, rato, e Arabidopsis thaliana genomas, bem como muitos outros.
resultados foram obtidos em duas plataformas de hardware: uma estação de trabalho desktop com processador Intel Core 2 2.4 GHz e 2 GB de RAM; e um servidor de grande memória com um processador AMD Opteron 2.4 GHz e 32 GB de RAM., Estes são denotados “PC” e “servidor”, respectivamente. Tanto o PC como o servidor rodam o Red Hat Enterprise Linux como release 4.
comparação com sabonete e Maq
Maq é um popular aliado que está entre as ferramentas de código aberto mais rápidas para alinhar milhões de leituras de ilumina ao genoma humano. SOAP é outra ferramenta de código aberto que foi relatada e usada em projetos de Leitura curta .
A Tabela 1 apresenta o desempenho e a sensibilidade do Bowtie v0.9.6, SOAP v1.10 e Maq v0.6.6. SOAP não poderia ser executado no PC porque a pegada de memória do SOAP excede a memória física do PC. O sabonete.,a versão contig do binário SOAP foi usada. Para comparação com o SOAP, Bowtie foi invocado com ‘-v 2’ para imitar a Política padrão de correspondência do SOAP (que permite até dois desfasamentos no alinhamento e desconsidera os valores de qualidade), e com ‘–maxns 5’ para simular a Política padrão do SOAP de filtrar leituras com cinco ou mais bases sem confiança. For the Maq comparison Bowtie is run with its default policy, which mimics Maq’s default policy of allowing up to two mismatches in the first 28 bases and enforcing an overall limit of 70 on the sum of the quality values at all mismatched read positions., Para tornar a pegada de memória de Bowtie mais comparável à de Maq, Bowtie é invocada com a opção ‘-z’ em todos os experimentos para garantir que apenas o índice forward ou mirror é residente em memória ao mesmo tempo.
O número de leituras alinhadas indica que sabão (67,3%) e Bowtie-v 2 (67,4%) têm sensibilidade comparável. Das leituras alinhadas por sabão ou Bowtie, 99,7% foram alinhados por ambos, 0,2% foram alinhados por Bowtie, mas não sabão, e 0,1% foram alinhados por sabão, mas não Bowtie. O Maq (74,7%) e o Bowtie (71,9%) têm também uma sensibilidade aproximadamente comparável, embora o Bowtie tenha um desfasamento de 2,8%., Das leituras alinhadas por Maq ou Bowtie, 96,0% foram alinhados por ambos, 0,1% foram alinhados por Bowtie mas não Maq, e 3,9% foram alinhados por Maq mas não Bowtie. Das leituras mapeadas por Maq mas não por Bowtie, quase todas são devidas a uma flexibilidade no algoritmo de alinhamento de Maq que permite que alguns alinhamentos tenham três mismatches na semente. O restante das leituras mapeadas por Maq, mas não Bowtie são devido ao teto de Bowtie de backtracking.a documentação de Maq menciona que ler contendo artefatos de poli-A pode prejudicar o desempenho de Maq., A tabela 2 apresenta desempenho e sensibilidade de Bowtie e Maq quando o conjunto de leitura é filtrado usando o comando ‘catfilter’do Maq para eliminar artefatos poly-A. O filtro elimina 438.145 de 8.839.010 leituras. Outros parâmetros experimentais são idênticos aos dos experimentos da Tabela 1, e as mesmas observações sobre a sensibilidade relativa de Bowtie e Maq aplicam-se aqui.
Lad length and performance
As sequencing technology improves, read lengths are growing beyond the 30-bp to 50-bp commonly seen in public databases today., Bowtie, Maq e SOAP suportam leituras de comprimentos até 1.024, 63, e 60 bp, respectivamente, e versões Maq 0.7.0 e mais tarde suportar comprimentos de leitura até 127 bp. A tabela 3 mostra os resultados de desempenho quando as três ferramentas são cada utilizada para alinhar três conjuntos de 2 M não aparadas lê, 36-bp, um conjunto de 50 bp e um conjunto de 76-bp conjunto, para o genoma humano na plataforma do servidor. Cada conjunto de 2 M é amostrado aleatoriamente de um conjunto maior (NCBI Short Read Archive: SRR003084 for 36-bp, SRR003092 for 50-bp, SRR003196 for 76-bp)., As leituras foram amostradas de modo que os três conjuntos de 2 M têm uma taxa de erro uniforme por base, calculada a partir de qualidades Phred por base. Todas as leituras passam pelo ‘catfilter’ do Maq.
Bowtie é executado tanto no seu modo padrão Maq como no seu modo de sabão ‘-v 2’. Bowtie também é dada a opção “- z ” para garantir que apenas o índice forward ou mirror é residente em memória ao mesmo tempo. Maq v0. 7. 1 foi usado em vez de Maq v0.6.6 para o conjunto de 76-bp porque v0.6.,6 não pode alinhar lê mais de 63 bp. SOAP não foi executado no conjunto 76-bp porque não suporta lê mais de 60 bp.
os resultados mostram que o algoritmo de Maq escala melhor em geral para comprimentos de leitura mais longos do que Bowtie ou sabonete. No entanto, o Bowtie no modo “-v 2 ” também balança muito bem. Bowtie em seu modo padrão Maq-like escalas bem de 36-bp a 50-bp lê, mas é substancialmente mais lento para leitura de 76-bp, embora ainda é mais do que uma ordem de magnitude mais rápido do que Maq.,
desempenho paralelo
alinhamento pode ser paralelizado distribuindo leituras através de linhas de pesquisa concorrentes. Bowtie permite ao usuário especificar um número desejado de threads (opção-p); Bowtie então lança o número especificado de threads usando a biblioteca pthreads. Os threads Bowtie sincronizam-se uns com os outros ao buscar leituras, resultados avançados, troca entre Índices, e executar várias formas de contabilidade global, como a marcação de uma leitura como ‘feito’., Caso contrário, threads são livres de operar em paralelo, acelerando substancialmente o alinhamento em computadores com núcleos de processador múltiplos. A imagem de memória do índice é compartilhada por todos os threads, e assim a pegada não aumenta substancialmente quando vários threads são usados. A tabela 4 mostra resultados de desempenho para executar Bowtie v0. 9. 6 no servidor de quatro núcleos com um, dois e quatro threads.,
Index building
Bowtie usa um algoritmo de indexação flexível que pode ser configurado para negociar entre a utilização da memória e o tempo de execução. O quadro 5 ilustra este compromisso ao indexar todo o genoma de referência humano (NCBI build 36.3, contigs). As corridas foram realizadas na plataforma do servidor. O indexador foi executado quatro vezes com diferentes limites superiores no uso da memória.,
O comunicado vezes comparam favoravelmente com o alinhamento vezes de competir ferramentas que realizam a indexação durante o alinhamento. Menos de 5 horas é necessário para Bowtie construir e consultar um índice humano inteiro com 8,84 milhões de leituras do projeto genoma 1000 (NCBI Short Read Archive:SRR001115) em um servidor, mais de seis vezes mais rápido do que a execução Maq equivalente., A linha inferior mais ilustra que o indexador de Bowtie, com argumentos apropriados, é eficiente o suficiente para executar em uma estação de trabalho típica com 2 GB de RAM. Ficheiro de dados adicionais 1 (discussões suplementares 3 e 4) explica o algoritmo e o conteúdo do índice resultante.
o Software
Bowtie é escrito em C++ e usa a biblioteca SeqAn . O conversor para o formato de mapeamento Maq usa o código do Maq.