Bowtie indeksuje Genom referencyjny za pomocą schematu opartego na transformacji Burrowsa-Wheelera (BWT) i indeksie FM . Indeks Bowtie dla ludzkiego genomu mieści się w 2.2 GB na dysku i ma ślad pamięci zaledwie 1.3 GB w czasie wyrównania, co pozwala na zapytanie na stacji roboczej z mniej niż 2 GB PAMIĘCI RAM.,
powszechną metodą wyszukiwania w indeksie FM jest algorytm dokładnego dopasowania Ferraginy i Manziniego . Bowtie nie przyjmuje po prostu tego algorytmu, ponieważ dokładne dopasowanie nie pozwala na błędy sekwencjonowania lub wariacje genetyczne. Wprowadzamy dwa nowe rozszerzenia, które sprawiają, że technika ma zastosowanie do wyrównywania krótkiego odczytu: świadomy jakości algorytm backtrackingu, który pozwala na niedopasowanie i sprzyja wyrównaniom wysokiej jakości; oraz „podwójne indeksowanie”, strategia mająca na celu uniknięcie nadmiernego backtrackingu., Bowtie aligner stosuje politykę podobną do Maq, ponieważ pozwala na niewielką liczbę niedopasowań w wysokiej jakości końcu każdego odczytu i umieszcza górną granicę sumy wartości jakości w niedopasowanych pozycjach wyrównania.
indeksowanie Burrowsa-Wheelera
BWT jest odwracalną permutacją znaków w tekście. Chociaż pierwotnie opracowany w kontekście kompresji danych, indeksowanie oparte na BWT pozwala na efektywne przeszukiwanie dużych tekstów w niewielkiej pamięci., Zastosowano go w zastosowaniach bioinformatycznych, w tym liczeniu oligomerów, wyrównaniu całego genomu, projektowaniu sond microarray i wyrównaniu Smith-Waterman do odniesienia wielkości człowieka .
transformacja Burrowsa-Wheelera tekstu T, BWT (T), jest skonstruowana w następujący sposób. Znak $ jest dołączany do T, gdzie $ nie jest w T i jest leksykograficznie mniejszy niż wszystkie znaki w T. macierz Burrowsa-Wheelera T jest skonstruowana jako macierz, której wiersze zawierają wszystkie cykliczne obroty t$. Wiersze są następnie sortowane leksykograficznie., BWT (T) jest sekwencją znaków w prawej kolumnie macierzy Burrowsa-Wheelera (rysunek 1a). BWT (T) ma taką samą długość jak oryginalny tekst T.
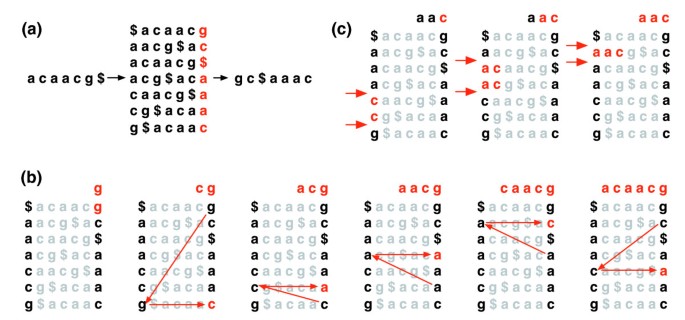
transformata Burrowsa-Wheelera. a) macierz Burrowsa-Wheelera i transformacja dla „acaacg”. b) kroki podjęte przez EXACTMATCH w celu określenia zakresu wierszy, a tym samym zestawu przyrostków odniesienia, poprzedzonych „aac”., (C) UNPERMUTE wielokrotnie stosuje Ostatnie pierwsze mapowanie (LF), aby odzyskać oryginalny tekst (czerwony w górnej linii) z przekształcenia Burrowsa-Wheelera (czarny w prawej kolumnie).
ta macierz ma właściwość o nazwie 'last first (LF) mapping'. I-te wystąpienie znaku X w ostatniej kolumnie odpowiada temu samemu znakowi tekstowemu, co i-te wystąpienie znaku X w pierwszej kolumnie. Ta właściwość leży u podstaw algorytmów wykorzystujących indeks BWT do nawigacji lub przeszukiwania tekstu., Rysunek 1b ilustruje UNPERMUTE, algorytm, który stosuje mapowanie LF wielokrotnie, aby odtworzyć T z BWT (T).
mapowanie LF jest również używane w dokładnym dopasowywaniu. Ponieważ macierz jest uporządkowana leksykograficznie, wiersze rozpoczynające się daną sekwencją pojawiają się kolejno. W szeregu kroków algorytm EXACTMATCH (rysunek 1c) oblicza zakres wierszy macierzy zaczynających się od kolejno dłuższych przyrostków zapytania. Na każdym kroku rozmiar zakresu kurczy się lub pozostaje taki sam., Po zakończeniu algorytmu wiersze rozpoczynające się od S0 (całe zapytanie) odpowiadają dokładnym wystąpieniom zapytania w tekście. Jeśli zakres jest pusty, tekst nie zawiera zapytania. UNPERMUTE jest przypisywany Burrowsowi i Wheelerowi, a dokładnie Ferraginie i Manzini . Zobacz dodatkowy plik danych 1 (dodatkowa dyskusja 1) po szczegóły.
Wyszukiwanie niedokładnych wyrównań
EXACTMATCH jest niewystarczające do wyrównania krótkiego odczytu, ponieważ wyrównania mogą zawierać niedopasowania, które mogą być spowodowane błędami sekwencjonowania, prawdziwymi różnicami między organizmami odniesienia i zapytań lub obiema., Wprowadzamy algorytm wyrównywania, który przeprowadza wyszukiwanie zwrotne, aby szybko znaleźć wyrównania spełniające określone zasady wyrównywania. Każdy znak w odczycie ma wartość liczbową jakości, z niższymi wartościami wskazującymi na większe prawdopodobieństwo błędu sekwencyjnego. Nasza polityka dopasowania pozwala na ograniczoną liczbę niedopasowań i preferuje dopasowania, w których suma wartości jakości we wszystkich niedopasowanych pozycjach jest niska.
wyszukiwanie przebiega podobnie do EXACTMATCH, obliczając zakresy macierzy dla kolejnych dłuższych przyrostków zapytania., Jeśli zakres staje się pusty (w tekście nie występuje przyrostek), algorytm może wybrać już dopasowaną pozycję zapytania i zastąpić tam inną bazę, wprowadzając niedopasowanie do wyrównania. Wyszukiwanie EXACTMATCH wznawia się zaraz po podstawionej pozycji. Algorytm wybiera tylko te podstawienia, które są zgodne z Polityką wyrównywania i które dają zmodyfikowany przyrostek, który występuje przynajmniej raz w tekście. Jeśli istnieje wiele kandydujących pozycji zastępczych, algorytm chciwie wybiera pozycję o minimalnej wartości jakości.,
scenariusze Backtrackingu rozgrywają się w kontekście struktury stosu, która rośnie po wprowadzeniu nowej substytucji i kurczy się, gdy aligner odrzuca wszystkie kandydujące wyrównania dla substytucji znajdujących się obecnie na stosie. Zob. Rysunek 2, aby zilustrować przebieg poszukiwań.
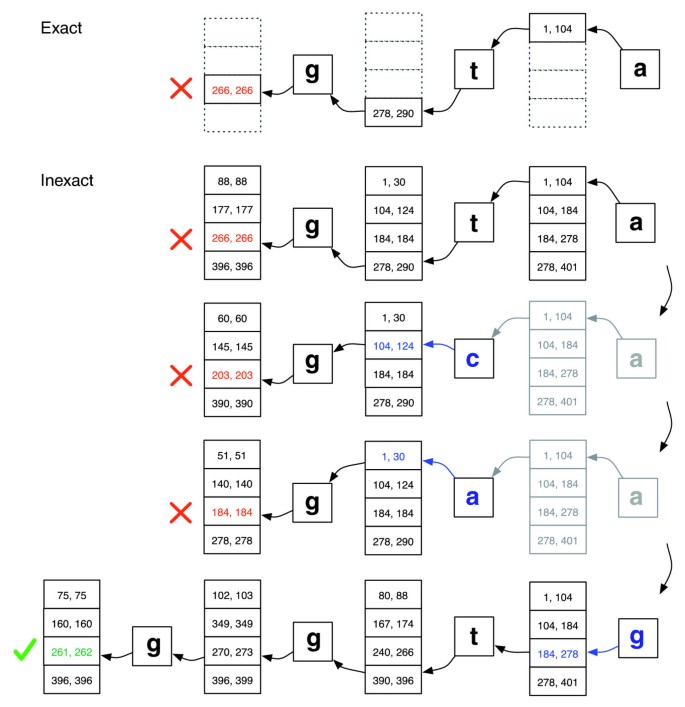
dokładne dopasowanie a niedokładne wyrównanie., Ilustracja jak exactmatch (góra) i Bowtie 's aligner (dół) postępują, gdy nie ma dokładnego dopasowania dla zapytania' ggta', ale istnieje wyrównanie jednego niedopasowania, gdy' a 'jest zastępowane przez’g'. Pary liczb zespolonych oznaczają zakresy wierszy macierzy zaczynające się od sufiksu obserwowanego do tego punktu. Czerwony znak X oznacza, gdzie algorytm napotka pusty zakres i albo przerywa (jak w exactmatch) lub backtracks (jak w algorytmie inexact). Zielony znacznik zaznacza, gdy algorytm znajdzie niepusty zakres ograniczający jedno lub więcej wystąpień raportowalnego wyrównania dla zapytania.,
W skrócie, Bowtie przeprowadza świadome jakości, chciwe, randomizowane, wyszukiwanie głębokości w przestrzeni możliwych wyrównań. Jeśli istnieje poprawne wyrównanie, Bowtie go znajdzie(z zastrzeżeniem pułapu backtrack omówionego w poniższej sekcji). Ponieważ wyszukiwanie jest chciwe, pierwsze poprawne wyrównanie napotkane przez Bowtie niekoniecznie będzie „najlepsze” pod względem liczby niedopasowań lub jakości., Użytkownik może polecić Bowtie kontynuowanie wyszukiwania, dopóki nie udowodni, że każde dopasowanie, które raportuje, jest 'najlepsze' pod względem liczby niedopasowań (używając opcji –best). Z naszego doświadczenia wynika, że tryb ten jest dwa do trzech razy wolniejszy niż tryb domyślny. Oczekujemy, że szybszy tryb domyślny będzie preferowany w przypadku dużych projektów ponownego uporządkowania.
użytkownik może również zdecydować się na raportowanie wszystkich wyrównań do określonej liczby (opcja-k) lub wszystkich wyrównań bez limitu liczby (opcja-a) dla danego odczytu., Jeśli w trakcie wyszukiwania Bowtie znajdzie N możliwych wyrównań dla danego zestawu podstawień, ale użytkownik zażądał tylko wyrównań K, gdzie k < n, Bowtie zgłosi K Z N wyrównań wybranych losowo. Zauważ, że te tryby mogą być znacznie wolniejsze niż domyślne. Z naszego doświadczenia wynika na przykład, że-k 1 jest ponad dwa razy szybszy od-k 2.
nadmierne cofanie
aligner, jak opisano do tej pory, może w niektórych przypadkach napotkać sekwencje powodujące nadmierne cofanie., Dzieje się tak, gdy aligner spędza większość swojego wysiłku bezowocnie cofając się do pozycji blisko 3′ końca zapytania. Bowtie łagodzi nadmierne cofanie dzięki nowatorskiej technice „podwójnego indeksowania”. Tworzone są dwa indeksy genomu: jeden zawierający BWT genomu, zwany indeksem „do przodu”, oraz drugi zawierający BWT genomu z jego sekwencją znaków odwróconą (nie odwróconą), zwaną indeksem „lustrem”. Aby zobaczyć, jak to pomaga, rozważ zasadę dopasowania, która pozwala na jedną niezgodność w wyrównaniu., Poprawne wyrównanie z jedną niezgodnością mieści się w jednym z dwóch przypadków, zgodnie z którymi połowa odczytu zawiera niezgodność. Bowtie przebiega w dwóch fazach odpowiadających tym dwóm przypadkom. Faza 1 ładuje indeks forward do pamięci i wywołuje aligner z ograniczeniem, którego nie może zastąpić w prawej połowie zapytania. Faza 2 wykorzystuje indeks lustrzany i wywołuje aligner na odwróconym zapytaniu, z ograniczeniem, którego aligner nie może zastąpić w pozycjach w prawej połowie odwróconego zapytania (w lewej połowie oryginalnego zapytania)., Ograniczenia dotyczące cofania do prawej połowy zapobiegają nadmiernemu cofaniu, podczas gdy zastosowanie dwóch faz i dwóch wskaźników zachowuje pełną czułość.
Niestety, nie jest możliwe uniknięcie nadmiernego cofania w pełni, gdy wyrównania mogą mieć dwa lub więcej niedopasowań. W naszych eksperymentach zauważyliśmy, że nadmierne cofanie jest znaczące tylko wtedy, gdy odczyt ma wiele pozycji niskiej jakości i nie wyrównuje się lub wyrównuje słabo do odniesienia., Przypadki te mogą wywołać ponad 200 backtrac ' ów na odczyt, ponieważ istnieje wiele kombinacji prawnych niskiej jakości pozycji, które należy zbadać, zanim wszystkie możliwości zostaną wyczerpane. Zmniejszamy ten koszt, wymuszając ograniczenie liczby dozwolonych ścieżek wstecznych przed zakończeniem wyszukiwania (domyślnie: 125). Limit ten uniemożliwia zgłaszanie pewnych legalnych, niskiej jakości dostosowań, ale oczekujemy, że jest to pożądany kompromis dla większości zastosowań.,
Wyszukiwanie stopniowe podobne do Maq
Bowtie pozwala użytkownikowi wybrać liczbę dozwolonych niedopasowań (domyślnie: dwa) w wysokiej jakości końcu odczytu (domyślnie: pierwsze 28 baz), a także maksymalną dopuszczalną odległość jakości ogólnego wyrównania (domyślnie: 70). Przyjmuje się, że wartości jakości są zgodne z definicją w PHRED, gdzie p jest prawdopodobieństwem błędu, A Q = – 10.
zarówno read, jak i jego odwrotne dopełnienie są kandydatami do wyrównania do odniesienia. Dla jasności w tej dyskusji uwzględniono jedynie orientację na przyszłość., Zobacz dodatkowy plik danych 1 (dyskusja dodatkowa 2), aby uzyskać wyjaśnienie, w jaki sposób odwrotne Uzupełnienie jest włączone.
pierwsze 28 podstaw na wysokiej jakości końcu odczytu nazywa się „nasionami”. Nasiona składają się z dwóch połówek: 14 bp na końcu wysokiej jakości (zwykle 5′) i 14 bp na końcu niskiej jakości, określanych odpowiednio jako „hi-half” I „lo-half”., Przy założeniu domyślnej polityki (dwa niedopasowania dozwolone w materiale siewnym), dostosowanie do celów sprawozdawczości będzie się składać w jednym z czterech przypadków: brak niedopasowania w materiale siewnym (przypadek 1); Brak niedopasowania w materiale siewnym, jeden lub dwa niedopasowania w materiale siewnym (przypadek 2); Brak niedopasowania w materiale siewnym, jeden lub dwa niedopasowania w materiale siewnym (przypadek 3); oraz jedno niedopasowanie w materiale siewnym, jedno niedopasowanie w materiale siewnym (przypadek 4).
wszystkie przypadki dopuszczają dowolną liczbę niedopasowań w części bezdźwięcznej odczytu, a wszystkie przypadki podlegają również ograniczeniu odległości jakościowej.,
algorytm Bowtie składa się z trzech faz, które naprzemiennie używają indeksów forward I mirror, jak pokazano na rysunku 3. Faza 1 wykorzystuje indeks lustrzany i wywołuje aligner, aby znaleźć wyrównania dla przypadków 1 i 2. Fazy 2 i 3 współpracują w celu znalezienia wyrównań dla przypadku 3: faza 2 znajduje częściowe wyrównania z niedopasowaniami tylko w połowie, a faza 3 próbuje rozszerzyć te częściowe wyrównania do pełnych wyrównań. Wreszcie, faza 3 wywołuje aligner, aby znaleźć wyrównania dla przypadku 4.,

trzy fazy algorytmu Bowtie dla polityki Maq-like. Podejście trójfazowe wyszukuje wyrównania dla dwóch przypadków niedopasowania od 1 do 4, minimalizując cofanie. Faza 1 wykorzystuje indeks lustrzany i wywołuje aligner, aby znaleźć wyrównania dla przypadków 1 i 2. Fazy 2 i 3 współpracują w celu znalezienia wyrównań dla przypadku 3: faza 2 znajduje częściowe wyrównania z niedopasowaniami tylko w połowie, a faza 3 próbuje rozszerzyć te częściowe wyrównania do pełnych wyrównań., Wreszcie, faza 3 wywołuje aligner, aby znaleźć wyrównania dla przypadku 4.
wyniki wydajności
oceniliśmy wydajność Bowtie za pomocą odczytów z 1000 genomów projektu pilotażowego (National Center for Biotechnology Information Short Read Archive:SRR001115). W sumie 8,84 miliona odczytów, około jednego pasa danych z instrumentu Illumina, zostało przyciętych do 35 bp i wyrównanych do ludzkiego genomu referencyjnego . O ile nie zaznaczono inaczej, odczytywane dane nie są filtrowane ani modyfikowane (poza przycinaniem) na podstawie sposobu ich wyświetlania w archiwum., Prowadzi to do około 70% do 75% odczytów dopasowujących się gdzieś do genomu. Z naszego doświadczenia wynika, że jest to typowe dla surowych danych z archiwum. Bardziej agresywne filtrowanie prowadzi do wyższych częstotliwości osiowania i szybszego osiowania.
wszystkie operacje wykonywane były na jednym procesorze. Bowtie speedupy zostały obliczone jako stosunek czasu ustawiania zegara ściennego. Zarówno zegar ścienny, jak i czas procesora są podane w celu wykazania, że obciążenie wejścia/wyjścia i niezgodność procesora nie są istotnymi czynnikami.
czas potrzebny na zbudowanie indeksu Bowtie nie został uwzględniony w czasie wykonywania Bowtie., W przeciwieństwie do konkurencyjnych narzędzi, Bowtie może ponownie użyć wstępnie obliczonego indeksu dla genomu odniesienia w wielu przebiegach wyrównania. Przewidujemy, że większość użytkowników po prostu pobierze takie indeksy z publicznego repozytorium. Strona Bowtie zawiera indeksy dla obecnych budów genomów człowieka, szympansa, myszy, psa, szczura i Arabidopsis thaliana, a także wielu innych.
wyniki uzyskano na dwóch platformach sprzętowych: desktopowej stacji roboczej z procesorem Intel Core 2 2,4 GHz i 2 GB PAMIĘCI RAM oraz wielkoformatowym serwerze z czterordzeniowym procesorem AMD Opteron 2,4 GHz i 32 GB PAMIĘCI RAM., Są one oznaczone odpowiednio ” PC ” i „server”. Zarówno na PC, jak i na serwerze działa Red Hat Enterprise Linux w wersji 4.
porównanie do SOAP i Maq
Maq jest popularnym alignerem, który jest jednym z najszybszych konkurencyjnych narzędzi open source do wyrównywania milionów odczytów Illumina do ludzkiego genomu. SOAP to kolejne narzędzie open source, które zostało zgłoszone i wykorzystane w projektach o krótkim czytaniu .
Tabela 1 przedstawia wydajność i czułość Bowtie v0.9.6, SOAP v1.10 i Maq v0.6.6. SOAP nie może być uruchomiony na komputerze, ponieważ ilość pamięci SOAP przekracza fizyczną pamięć komputera. Mydło.,zastosowano wersję binarną mydła contiga. Dla porównania z SOAP, Bowtie został wywołany za pomocą '- v 2′, aby naśladować domyślną politykę dopasowania SOAP (która pozwala na maksymalnie dwa niedopasowania w wyrównaniu i ignoruje wartości jakości), oraz za pomocą '–maxns 5′, aby symulować domyślną politykę filtrowania odczytów SOAP z pięcioma lub więcej bazami no-confidence. Dla porównania MAQ Bowtie jest uruchamiany z jego domyślną polityką, która naśladuje domyślną politykę MAQ dopuszczającą do dwóch niedopasowań w pierwszych 28 bazach i wymuszającą ogólny limit 70 na sumie wartości jakości we wszystkich niedopasowanych pozycjach odczytu., Aby footprint pamięci Bowtie jest bardziej porównywalny do Maq, Bowtie jest wywoływany z opcją „- z ” we wszystkich eksperymentach, aby zapewnić, że tylko indeks forward lub mirror jest rezydentny w pamięci w tym samym czasie.
Liczba odczytów wskazuje, że SOAP (67,3%) i Bowtie-v 2 (67,4%) mają porównywalną czułość. Z odczytów wyrównanych przez SOAP lub Bowtie, 99,7% było wyrównanych przez oba, 0,2% było wyrównane przez Bowtie, ale nie SOAP, a 0,1% było wyrównane przez SOAP, ale nie Bowtie. Maq (74,7%) i Bowtie (71,9%) również mają mniej więcej porównywalną czułość, chociaż Bowtie pozostaje o 2,8%., Z odczytów wyrównanych przez Maq lub Bowtie, 96,0% było wyrównanych przez oba, 0,1% było wyrównane przez Bowtie, ale nie Maq, a 3,9% było wyrównane przez Maq, ale nie Bowtie. Z odczytów mapowanych przez Maq, ale nie Bowtie, prawie wszystkie są spowodowane elastycznością algorytmu wyrównywania Maq, który pozwala niektórym wyrównaniom mieć trzy niedopasowania w zalążku. Pozostałe odczyty mapowane przez Maq, ale nie Bowtie są spowodowane pułapem cofania Bowtie.
dokumentacja Maq wspomina, że odczyty zawierające „artefakty poly-A” mogą negatywnie wpływać na wydajność Maq., Tabela 2 przedstawia wydajność i czułość Bowtie i Maq, gdy zestaw odczytu jest filtrowany za pomocą polecenia MAQ 'catfilter' w celu wyeliminowania artefaktów poly-A. Filtr eliminuje 438,145 z 8,839,010 odsłon. Inne parametry eksperymentalne są identyczne z tymi z eksperymentów w tabeli 1, i te same obserwacje dotyczące względnej czułości Bowtie i Maq stosuje się tutaj.
Długość odczytu i wydajność
wraz z poprawą technologii sekwencjonowania, długość odczytu rośnie poza 30-bp do 50-bp powszechnie spotykanych obecnie w publicznych bazach danych., Bowtie, Maq i SOAP obsługują odczyty długości do odpowiednio 1,024, 63 i 60 bp, a wersje Maq 0.7.0 i późniejsze obsługują odczyty długości do 127 bp. Tabela 3 pokazuje wyniki wydajności, gdy trzy narzędzia są używane do wyrównania trzech zestawów 2 M untrimmed odczytów, Zestaw 36-bp, zestaw 50-bp i zestaw 76-bp, do ludzkiego genomu na platformie serwerowej. Każdy zestaw 2 M jest losowo pobierany z większego zestawu (NCBI Short Read Archive: SRR003084 dla 36-bp, SRR003092 dla 50-bp, SRR003196 dla 76-bp)., Odczyty próbkowano w taki sposób, że trzy zestawy 2 M mają jednolity poziom błędu dla podstawy, obliczony na podstawie jakości Phred dla podstawy. Wszystkie odczyty przechodzą przez „catfilter” Maq.
Bowtie jest uruchamiany zarówno w domyślnym trybie MAQ, jak i w trybie SOAP '-V 2′. Bowtie otrzymuje również opcję „- z”, aby upewnić się, że tylko indeks forward lub mirror jest rezydentny w pamięci w tym samym czasie. Maq v0.7.1 został użyty zamiast Maq v0. 6. 6 dla zestawu 76-bp, ponieważ v0. 6.,6 nie może wyrównać odczytów dłuższych niż 63 bp. SOAP nie był uruchamiany na zestawie 76-bp, ponieważ nie obsługuje odczytów dłuższych niż 60 bp.
wyniki pokazują, że algorytm Maq skaluje się lepiej ogólnie do dłuższych długości odczytu niż Bowtie czy SOAP. Jednak Bowtie w trybie SOAP-like '- v 2 ' również skaluje się bardzo dobrze. Bowtie w domyślnym trybie Maq-like skaluje się dobrze od odczytów 36-bp do 50-bp, ale jest znacznie wolniejszy dla odczytów 76-bp, chociaż nadal jest o rząd wielkości szybszy niż Maq.,
wydajność równoległa
wyrównanie może być równoległe poprzez dystrybucję odczytów między współbieżnymi wątkami wyszukiwania. Bowtie pozwala użytkownikowi określić żądaną liczbę wątków( opcja-p); Bowtie uruchamia określoną liczbę wątków za pomocą biblioteki pthreads. Wątki Bowtie synchronizują się ze sobą podczas pobierania odczytów, wysyłania wyników, przełączania między indeksami i wykonywania różnych form globalnej księgowości, takich jak oznaczanie odczytu jako „gotowe”., W przeciwnym razie wątki mogą działać równolegle, znacznie przyspieszając wyrównanie na komputerach z wieloma rdzeniami procesora. Obraz pamięci indeksu jest współdzielony przez wszystkie wątki, więc footprint nie zwiększa się znacząco, gdy używa się wielu wątków. Tabela 4 pokazuje wyniki wydajności dla uruchomienia Bowtie v0. 9. 6 na serwerze czterordzeniowym z jednym, dwoma i czterema wątkami.,
Budynek indeks
Bowtie wykorzystuje elastyczny algorytm indeksowania, które mogą być skonfigurowane do handlu off między wykorzystaniem pamięci i czasu pracy. Tabela 5 ilustruje ten kompromis podczas indeksowania całego ludzkiego genomu referencyjnego (NCBI build 36.3, contigs). Uruchamianie odbywało się na platformie serwerowej. Indekser był uruchamiany cztery razy z różnymi wyższymi limitami zużycia pamięci.,
Podane czasy są korzystnie porównywane z czasami wyrównywania w konkurencyjnych narzędziach, które wykonują indeksowanie podczas wyrównywania. Mniej niż 5 godzin jest wymagane dla Bowtie zarówno zbudować i zapytanie całego ludzkiego indeksu z 8.84 mln odczytów z projektu 1000 genomu (NCBI Short Read Archive: SRR001115) na serwerze, ponad sześciokrotnie szybciej niż równoważny MAQ uruchomić., Najniższy wiersz pokazuje, że Bowtie indexer, z odpowiednimi argumentami, jest wystarczająco wydajny, aby działać na typowej stacji roboczej z 2 GB PAMIĘCI RAM. Dodatkowy plik danych 1 (dodatkowe dyskusje 3 i 4) wyjaśnia algorytm i zawartość wynikowego indeksu.
oprogramowanie
Bowtie jest napisane w C++ i korzysta z biblioteki SeqAn . Konwerter do formatu mapowania Maq wykorzystuje kod z Maq.