Bowtie indicizza il genoma di riferimento utilizzando uno schema basato sulla trasformazione di Burrows-Wheeler (BWT) e l’indice FM . Un indice Bowtie per il genoma umano si adatta a 2,2 GB su disco e ha un ingombro di memoria di appena 1,3 GB al momento dell’allineamento, permettendogli di essere interrogato su una workstation con meno di 2 GB di RAM.,
Il metodo comune per la ricerca in un indice FM è l’algoritmo di corrispondenza esatta di Ferragina e Manzini . Bowtie non adotta semplicemente questo algoritmo perché la corrispondenza esatta non consente errori di sequenziamento o variazioni genetiche. Introduciamo due nuove estensioni che rendono la tecnica applicabile all’allineamento a lettura breve: un algoritmo di backtracking consapevole della qualità che consente disallineamenti e favorisce allineamenti di alta qualità; e ‘double indexing’, una strategia per evitare un eccessivo backtracking., L’allineatore Bowtie segue una politica simile a Maq, in quanto consente un piccolo numero di disallineamenti all’interno della fine di alta qualità di ogni lettura, e pone un limite superiore alla somma dei valori di qualità in posizioni di allineamento non corrispondenti.
Burrows-Wheeler indicizzazione
Il BWT è una permutazione reversibile dei caratteri in un testo. Sebbene originariamente sviluppato nel contesto della compressione dei dati, l’indicizzazione basata su BWT consente di cercare in modo efficiente testi di grandi dimensioni in un ingombro di memoria ridotto., È stato applicato alle applicazioni di bioinformatica, compreso il conteggio degli oligomeri , l’allineamento del intero genoma , piastrellando la progettazione della sonda del microarray e l’allineamento di Smith-Waterman ad un riferimento a misura d’uomo .
La trasformazione Burrows-Wheeler di un testo T, BWT(T), è costruita come segue. Il carattere $ è aggiunto a T, dove $ non è in T ed è lessicograficamente inferiore a tutti i caratteri in T. La matrice Burrows-Wheeler di T è costruita come la matrice le cui righe comprendono tutte le rotazioni cicliche di T$. Le righe vengono quindi ordinate lessicograficamente., BWT (T) è la sequenza di caratteri nella colonna più a destra della matrice Burrows-Wheeler (Figura 1a). BWT (T) ha la stessa lunghezza del testo originale T.
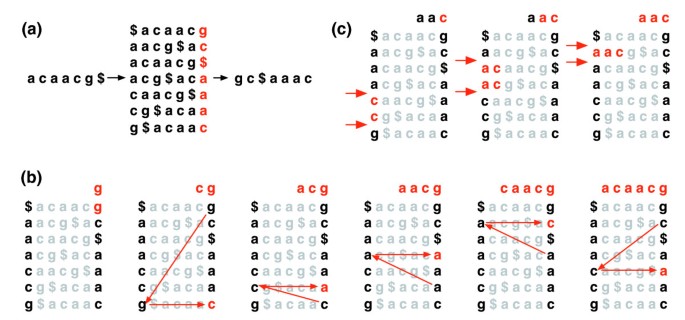
Burrows-Wheeler transform. a) La matrice e la trasformazione di Burrows-Wheeler per “acaacg”. (b) Misure adottate da EXACTMATCH per identificare l’intervallo di righe, e quindi l’insieme di suffissi di riferimento, preceduto da ‘aac’., (c) UNPERMUTE applica ripetutamente l’ultima mappatura prima (LF) per recuperare il testo originale (in rosso sulla riga superiore) dalla trasformazione Burrows-Wheeler (in nero nella colonna più a destra).
Questa matrice ha una proprietà chiamata ‘last first (LF) mapping’. L’ith occorrenza del carattere X nell’ultima colonna corrisponde allo stesso carattere di testo dell’ith occorrenza di X nella prima colonna. Questa proprietà è alla base degli algoritmi che utilizzano l’indice BWT per navigare o cercare il testo., La figura 1b illustra UNPERMUTE, un algoritmo che applica ripetutamente la mappatura LF per ricreare T da BWT (T).
La mappatura LF viene utilizzata anche nella corrispondenza esatta. Poiché la matrice è ordinata lessicograficamente, le righe che iniziano con una determinata sequenza appaiono consecutivamente. In una serie di passaggi, l’algoritmo EXACTMATCH (Figura 1c) calcola l’intervallo di righe della matrice che iniziano con suffissi successivamente più lunghi della query. Ad ogni passaggio, la dimensione dell’intervallo si restringe o rimane la stessa., Al termine dell’algoritmo, le righe che iniziano con S0 (l’intera query) corrispondono alle occorrenze esatte della query nel testo. Se l’intervallo è vuoto, il testo non contiene la query. UNPERMUTE è attribuibile a Burrows e Wheeler e EXACTMATCH a Ferragina e Manzini . Vedere File di dati aggiuntivi 1 (Discussione supplementare 1) per i dettagli.
La ricerca di allineamenti inesatti
EXACTMATCH è insufficiente per l’allineamento di lettura breve perché gli allineamenti possono contenere disallineamenti, che possono essere dovuti a errori di sequenziamento, differenze autentiche tra organismi di riferimento e query o entrambi., Introduciamo un algoritmo di allineamento che esegue una ricerca di backtracking per trovare rapidamente allineamenti che soddisfano un criterio di allineamento specificato. Ogni carattere in una lettura ha un valore numerico di qualità, con valori più bassi che indicano una maggiore probabilità di un errore di sequenziamento. La nostra politica di allineamento consente un numero limitato di disallineamenti e preferisce allineamenti in cui la somma dei valori di qualità in tutte le posizioni non corrispondenti è bassa.
La ricerca procede in modo simile a EXACTMATCH, calcolando intervalli di matrici per suffissi di query successivamente più lunghi., Se l’intervallo diventa vuoto (un suffisso non si verifica nel testo), l’algoritmo può selezionare una posizione di query già abbinata e sostituire una base diversa, introducendo una mancata corrispondenza nell’allineamento. La ricerca EXACTMATCH riprende da subito dopo la posizione sostituita. L’algoritmo seleziona solo le sostituzioni che sono coerenti con la politica di allineamento e che producono un suffisso modificato che si verifica almeno una volta nel testo. Se ci sono più posizioni di sostituzione dei candidati, l’algoritmo seleziona avidamente una posizione con un valore di qualità minimo.,
Gli scenari di backtracking si svolgono nel contesto di una struttura dello stack che cresce quando viene introdotta una nuova sostituzione e si restringe quando l’allineatore rifiuta tutti gli allineamenti candidati per le sostituzioni attualmente nello stack. Vedere Figura 2 per un’illustrazione di come potrebbe procedere la ricerca.
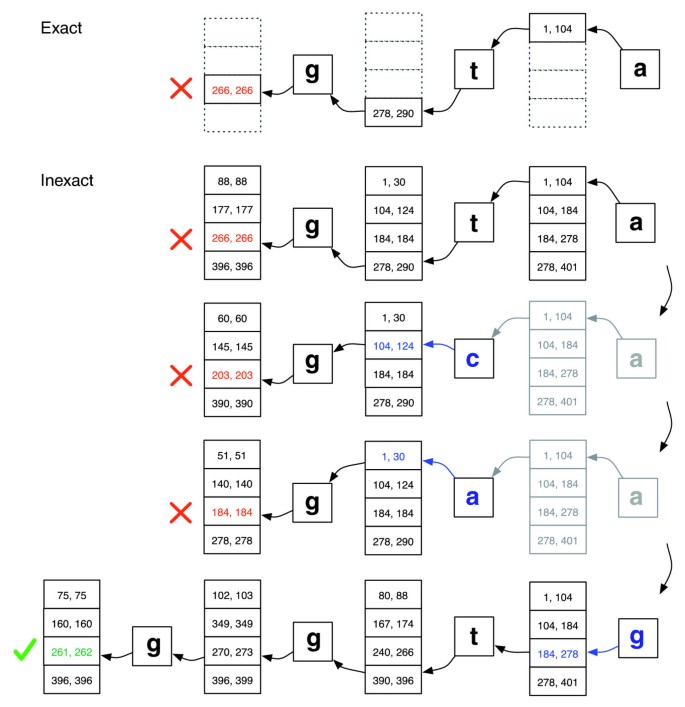
Corrispondenza esatta contro allineamento inesatto., Illustrazione di come EXACTMATCH (in alto) e l’allineamento di Bowtie (in basso) procedono quando non c’è corrispondenza esatta per la query ‘ggta’ ma c’è un allineamento di una mancata corrispondenza quando ‘a’ è sostituito da ‘g’. Coppie di numeri in scatola denotano intervalli di righe di matrice che iniziano con il suffisso osservato fino a quel punto. Una X rossa indica dove l’algoritmo incontra un intervallo vuoto e abortisce (come in EXACTMATCH) o backtracks (come nell’algoritmo inexact). Un segno di spunta verde indica dove l’algoritmo trova un intervallo non vuoto che delimita una o più occorrenze di un allineamento segnalabile per la query.,
In breve, Bowtie conduce una ricerca basata sulla qualità, avida, randomizzata e approfondita attraverso lo spazio dei possibili allineamenti. Se esiste un allineamento valido, allora Bowtie lo troverà (soggetto al soffitto backtrack discusso nella sezione seguente). Poiché la ricerca è avida, il primo allineamento valido incontrato da Bowtie non sarà necessariamente il “migliore” in termini di numero di disallineamenti o in termini di qualità., L’utente può istruire Bowtie a continuare la ricerca finché non può dimostrare che qualsiasi allineamento riportato è “migliore” in termini di numero di disallineamenti (usando l’opzione best best). Nella nostra esperienza, questa modalità è da due a tre volte più lenta della modalità predefinita. Ci aspettiamo che la modalità predefinita più veloce sarà preferito per grandi progetti di ri-sequenziamento.
L’utente può anche optare per Bowtie per segnalare tutti gli allineamenti fino a un numero specificato (opzione-k) o tutti gli allineamenti senza limiti sul numero (opzione-a) per una data lettura., Se nel corso della sua ricerca Bowtie trova N possibili allineamenti per un dato insieme di sostituzioni, ma l’utente ha richiesto solo allineamenti K dove K < N, Bowtie riporterà K degli allineamenti N selezionati a caso. Nota che queste modalità possono essere molto più lente di quelle predefinite. Nella nostra esperienza, ad esempio,- k 1 è più del doppio di-k 2.
Backtracking eccessivo
L’allineatore come descritto finora può, in alcuni casi, incontrare sequenze che causano un backtracking eccessivo., Ciò si verifica quando l’allineatore spende la maggior parte del suo sforzo inutilmente backtracking in posizioni vicine alla fine 3′ della query. Bowtie mitiga eccessivo backtracking con la nuova tecnica di ‘doppia indicizzazione’. Vengono creati due indici del genoma: uno contenente il BWT del genoma, chiamato indice’ forward’, e un secondo contenente il BWT del genoma con la sua sequenza di caratteri invertita (non inversa completata) chiamato indice’ mirror’. Per vedere come questo aiuta, prendere in considerazione un criterio di corrispondenza che consente una mancata corrispondenza nel tracciato., Un allineamento valido con una mancata corrispondenza rientra in uno dei due casi in base ai quali metà della lettura contiene la mancata corrispondenza. Bowtie procede in due fasi corrispondenti a questi due casi. La fase 1 carica l’indice forward in memoria e richiama l’allineatore con il vincolo che non può sostituire nelle posizioni nella metà destra della query. La fase 2 utilizza l’indice mirror e richiama l’aligner sulla query invertita, con il vincolo che l’aligner non può sostituire nelle posizioni nella metà destra della query invertita (metà sinistra della query originale)., I vincoli sul backtracking nella metà destra impediscono un eccessivo backtracking, mentre l’uso di due fasi e due indici mantiene la piena sensibilità.
Sfortunatamente, non è possibile evitare un backtracking eccessivo quando gli allineamenti sono autorizzati ad avere due o più disallineamenti. Nei nostri esperimenti, abbiamo osservato che il backtracking eccessivo è significativo solo quando una lettura ha molte posizioni di bassa qualità e non si allinea o si allinea male al riferimento., Questi casi possono innescare oltre 200 backtracks per lettura perché ci sono molte combinazioni legali di posizioni di bassa qualità da esplorare prima che tutte le possibilità siano esaurite. Attenuiamo questo costo applicando un limite al numero di backtracks consentiti prima che una ricerca venga terminata (default: 125). Il limite impedisce che vengano segnalati alcuni allineamenti legittimi e di bassa qualità, ma ci aspettiamo che questo sia un compromesso auspicabile per la maggior parte delle applicazioni.,
Phased Maq-like search
Bowtie consente all’utente di selezionare il numero di disallineamenti consentiti (default: due) nella fine di alta qualità di una lettura (default: le prime 28 basi) così come la distanza massima qualità accettabile dell’allineamento complessivo (default: 70). Si presume che i valori di qualità seguano la definizione in PHRED, dove p è la probabilità di errore e Q = -10log p.
Sia la lettura che il suo complemento inverso sono candidati per l’allineamento al riferimento. Per chiarezza, questa discussione considera solo l’orientamento in avanti., Vedere il file di dati aggiuntivo 1 (Discussione supplementare 2) per una spiegazione di come è incorporato il complemento inverso.
Le prime 28 basi sull’estremità di alta qualità della lettura sono definite “seme”. Il seme consiste di due metà: 14 bp sull’estremità di alta qualità (solitamente l’estremità 5′) e 14 bp sull’estremità di bassa qualità, denominate rispettivamente “hi-half” e “lo-half”., Supponendo la politica predefinita (due disallineamenti consentiti nel seme), un allineamento segnalabile cadrà in uno dei quattro casi: nessun disallineamento nel seme (caso 1); nessun disallineamento in hi-half, uno o due disallineamenti in lo-half (caso 2); nessun disallineamento in lo-half, uno o due disallineamenti in hi-half (caso 3); e un disallineamento in hi-half, un disallineamento in lo-half (caso 4).
Tutti i casi consentono un numero qualsiasi di disallineamenti nella parte nonseed della lettura e tutti i casi sono anche soggetti al vincolo di distanza di qualità.,
L’algoritmo Bowtie consiste in tre fasi che si alternano tra l’utilizzo degli indici forward e mirror, come illustrato nella Figura 3. La fase 1 utilizza l’indice mirror e richiama l’allineatore per trovare gli allineamenti per i casi 1 e 2. Le fasi 2 e 3 cooperano per trovare allineamenti per il caso 3: la fase 2 trova allineamenti parziali con disallineamenti solo nell’hi-half e la fase 3 tenta di estendere tali allineamenti parziali in allineamenti completi. Infine, la fase 3 richiama l’allineatore per trovare gli allineamenti per il caso 4.,

Le tre fasi dell’algoritmo Bowtie per la politica simile a Maq. Un approccio trifase trova allineamenti per due casi di mancata corrispondenza da 1 a 4 riducendo al minimo il backtracking. La fase 1 utilizza l’indice mirror e richiama l’allineatore per trovare gli allineamenti per i casi 1 e 2. Le fasi 2 e 3 cooperano per trovare allineamenti per il caso 3: la fase 2 trova allineamenti parziali con disallineamenti solo nell’hi-half e la fase 3 tenta di estendere tali allineamenti parziali in allineamenti completi., Infine, la fase 3 richiama l’allineatore per trovare gli allineamenti per il caso 4.
Risultati delle prestazioni
Abbiamo valutato le prestazioni di Bowtie utilizzando letture dal progetto pilota 1,000 Genomi (National Center for Biotechnology Information Short Read Archive:SRR001115). Un totale di 8,84 milioni di letture, circa una corsia di dati da uno strumento Illumina, sono stati tagliati a 35 bp e allineati al genoma umano di riferimento . Se non diversamente specificato, i dati letti non vengono filtrati o modificati (oltre al ritaglio) dal modo in cui appaiono nell’archivio., Questo porta a circa il 70% al 75% delle letture allineando da qualche parte al genoma. Nella nostra esperienza, questo è tipico per i dati grezzi dall’archivio. Un filtraggio più aggressivo porta a tassi di allineamento più elevati e un allineamento più veloce.
Tutte le esecuzioni sono state eseguite su una singola CPU. Gli speedup di Bowtie sono stati calcolati come un rapporto tra i tempi di allineamento dell’orologio da parete. Sia l’orologio da parete che i tempi della CPU sono dati per dimostrare che il carico di input/output e la contesa della CPU non sono fattori significativi.
Il tempo necessario per costruire l’indice Bowtie non è stato incluso nei tempi di esecuzione Bowtie., A differenza di strumenti concorrenti, Bowtie può riutilizzare un indice pre-calcolato per il genoma di riferimento in molte piste di allineamento. Prevediamo che la maggior parte degli utenti scaricherà semplicemente tali indici da un repository pubblico. Il sito Bowtie fornisce indici per le attuali build dei genomi umani, scimpanzé, topo, cane, ratto e Arabidopsis thaliana, così come molti altri.
I risultati sono stati ottenuti su due piattaforme hardware: una workstation desktop con processore Intel Core 2 da 2,4 GHz e 2 GB di RAM; e un server di grande memoria con un processore AMD Opteron da 2,4 GHz a quattro core e 32 GB di RAM., Questi sono indicati rispettivamente ‘ PC ‘e’ server’. Sia il PC che il server eseguono Red Hat Enterprise Linux COME versione 4.
Confronto con SOAP e Maq
Maq è un allineatore popolare che è tra i più veloci strumenti open source concorrenti per allineare milioni di Illumina legge al genoma umano. SOAP è un altro strumento open source che è stato segnalato e utilizzato in progetti di breve lettura .
La tabella 1 presenta le prestazioni e la sensibilità di Bowtie v0.9.6, SOAP v1.10 e Maq v0.6.6. Impossibile eseguire SOAP sul PC perché l’ingombro di memoria di SOAP supera la memoria fisica del PC. Il sapone.,è stata utilizzata la versione di contig del binario SOAP. Per il confronto con SOAP, Bowtie è stato invocato con ‘ – v 2 ‘per imitare la politica di corrispondenza predefinita di SOAP (che consente fino a due disallineamenti nell’allineamento e ignora i valori di qualità) e con’ max maxns 5 ‘ per simulare la politica predefinita di SOAP di filtrare le letture con cinque o più basi di sfiducia. Per il confronto Maq, Bowtie viene eseguito con la sua politica predefinita, che imita la politica predefinita di Maq di consentire fino a due disallineamenti nelle prime 28 basi e di imporre un limite complessivo di 70 sulla somma dei valori di qualità in tutte le posizioni di lettura non corrispondenti., Per rendere l’impronta di memoria di Bowtie più paragonabile a quella di Maq, Bowtie viene invocato con l’opzione’ – z ‘ in tutti gli esperimenti per garantire che solo l’indice forward o mirror sia residente in memoria contemporaneamente.
Il numero di letture allineate indica che SOAP (67,3%) e Bowtie-v 2 (67,4%) hanno sensibilità comparabili. Delle letture allineate da SOAP o Bowtie, il 99,7% era allineato da entrambi, lo 0,2% era allineato da Bowtie ma non da SOAP e lo 0,1% era allineato da SOAP ma non da Bowtie. Anche Maq (74,7%) e Bowtie (71,9%) hanno una sensibilità approssimativamente paragonabile, sebbene Bowtie sia in ritardo del 2,8%., Tra le letture allineate da Maq o Bowtie, il 96,0% era allineato da entrambi, lo 0,1% era allineato da Bowtie ma non da Maq, e il 3,9% era allineato da Maq ma non da Bowtie. Delle letture mappate da Maq ma non da Bowtie, quasi tutte sono dovute a una flessibilità nell’algoritmo di allineamento di Maq che consente ad alcuni allineamenti di avere tre disallineamenti nel seme. Il resto delle letture mappate da Maq ma non Bowtie sono dovute al soffitto backtracking di Bowtie.
La documentazione di Maq menziona che le letture contenenti “artefatti poly-A” possono compromettere le prestazioni di Maq., La Tabella 2 presenta le prestazioni e la sensibilità di Bowtie e Maq quando il set di lettura viene filtrato utilizzando il comando ‘catfilter’ di Maq per eliminare gli artefatti poly-A. Il filtro elimina 438.145 su 8.839.010 letture. Altri parametri sperimentali sono identici a quelli degli esperimenti nella Tabella 1, e le stesse osservazioni sulla sensibilità relativa di Bowtie e Maq si applicano qui.
Lunghezza e prestazioni di lettura
Man mano che la tecnologia di sequenziamento migliora, le lunghezze di lettura stanno crescendo oltre i 30-bp a 50-bp comunemente visti nei database pubblici oggi., Bowtie, Maq e SOAP supportano letture di lunghezze fino a 1.024, 63 e 60 bp, rispettivamente, e le versioni Maq 0.7.0 e successive supportano lunghezze di lettura fino a 127 bp. La tabella 3 mostra i risultati delle prestazioni quando i tre strumenti vengono utilizzati ciascuno per allineare tre serie di letture non tagliate di 2 M, un set di 36 bp, un set di 50 bp e un set di 76 bp, al genoma umano sulla piattaforma server. Ogni set di 2 M è casualmente campionato da un set più grande (NCBI breve Lettura Archivio: SRR003084 per 36-bp, SRR003092 per 50-bp, SRR003196 per 76-bp)., Le letture sono state campionate in modo tale che le tre serie di 2 M abbiano un tasso di errore uniforme per base, calcolato in base alle qualità Phred per base. Tutte le letture passano attraverso il “catfilter” di Maq.
Bowtie viene eseguito sia nella modalità predefinita simile a Maq che nella modalità SOAP ‘-v 2′. Bowtie è anche dato il’ – z ‘ opzione per garantire che solo il forward o mirror index è residente in memoria in una sola volta. Maq v0.7.1 è stato utilizzato al posto di Maq v0.6.6 per il set 76-bp perché v0. 6.,6 impossibile allineare le letture più lunghe di 63 bp. SOAP non è stato eseguito sul set 76-bp perché non supporta letture più lunghe di 60 bp.
I risultati mostrano che l’algoritmo di Maq scala meglio in generale a lunghezze di lettura più lunghe rispetto a Bowtie o SOAP. Tuttavia, Bowtie in modalità SOAP-like ‘- v 2 ‘ scala anche molto bene. Bowtie nella sua modalità predefinita Maq-like scala bene da 36-bp a 50-bp legge, ma è sostanzialmente più lento per 76-bp legge, anche se è ancora più di un ordine di grandezza più veloce di Maq.,
Prestazioni parallele
L’allineamento può essere parallelizzato distribuendo le letture tra thread di ricerca simultanei. Bowtie consente all’utente di specificare un numero desiderato di thread (opzione-p); Bowtie avvia quindi il numero specificato di thread utilizzando la libreria pthreads. I thread Bowtie si sincronizzano tra loro durante il recupero delle letture, l’output dei risultati, il passaggio da un indice all’altro e l’esecuzione di varie forme di contabilità globale, come contrassegnare una lettura come “fatta”., In caso contrario, i thread sono liberi di operare in parallelo, accelerando sostanzialmente l’allineamento su computer con più core del processore. L’immagine di memoria dell’indice è condivisa da tutti i thread e quindi l’impronta non aumenta sostanzialmente quando vengono utilizzati più thread. La tabella 4 mostra i risultati delle prestazioni per l’esecuzione di Bowtie v0.9. 6 sul server a quattro core con uno, due e quattro thread.,
Index building
Bowtie utilizza un algoritmo di indicizzazione flessibile che può essere configurato per scambiare tra l’utilizzo della memoria e il tempo di esecuzione. La tabella 5 illustra questo compromesso quando si indicizza l’intero genoma umano di riferimento (NCBI build 36.3, contigs). Le esecuzioni sono state eseguite sulla piattaforma server. L’indicizzatore è stato eseguito quattro volte con diversi limiti superiori sull’utilizzo della memoria.,
I tempi riportati si confrontano favorevolmente con i tempi di allineamento degli strumenti concorrenti che eseguono l’indicizzazione durante l’allineamento. Meno di 5 ore è necessario per Bowtie sia per costruire e interrogare un indice intero umano con 8,84 milioni di letture dal progetto genoma 1,000 (NCBI breve Read Archive: SRR001115) su un server, più di sei volte più veloce rispetto alla corsa Maq equivalente., La riga più in basso illustra che l’indicizzatore Bowtie, con argomenti appropriati, è abbastanza efficiente in memoria da funzionare su una tipica workstation con 2 GB di RAM. File di dati aggiuntivi 1 (Discussioni supplementari 3 e 4) spiega l’algoritmo e il contenuto dell’indice risultante.
Software
Bowtie è scritto in C++ e utilizza la libreria SeqAn . Il convertitore nel formato di mappatura Maq utilizza il codice da Maq.